Del Enfermedad de andersen es una forma particularmente grave de enfermedad por almacenamiento de glucógeno, una enfermedad hereditaria caracterizada por la formación de glucógeno anormal. El pronóstico de la enfermedad es muy malo.
¿Qué es la enfermedad de Andersen?

© ag visuell - stock.adobe.com
En el contexto de Enfermedad de andersen se almacena una forma inusual de glucógeno. Este glucógeno tiene una estructura similar a la amilopectina, un alto porcentaje de la cual se encuentra en el almidón vegetal. Por lo general, el glucógeno está muy ramificado. En la enfermedad de Andersen, sin embargo, solo hay un polisacárido ramificado débilmente.
La enfermedad se caracteriza por un rápido agrandamiento del hígado, que rápidamente conduce a una cirrosis hepática. El polisacárido anormal ya no se puede descomponer y continúa acumulándose. La deficiencia o incluso la falta de la enzima amilo-1,4-1,6-transglucosidasa es responsable de la formación defectuosa de glucógeno. Proporciona la ramificación en esta molécula de polisacárido.
La enfermedad es muy rara, pero todavía se presenta en diversas formas o formas. En la forma extremadamente grave, el niño a menudo nace muerto. También se han descrito formas más ligeras que comienzan a una edad posterior. En cualquier caso, sin embargo, existe una mutación en el gen (GBE1), que se encuentra en el cromosoma 3.
causas
La causa de la enfermedad de Andersen es un defecto genético en el gen GBE1 en el cromosoma 3, que puede heredarse como un rasgo autosómico recesivo. Este gen es responsable de la síntesis de la enzima amilo-1,4-1,6-transglucosidasa. Si falta esta enzima o si tiene una funcionalidad limitada, ya no se puede sintetizar glucógeno normal. La enzima es responsable de la ramificación de la molécula de polisacarosa.
Si esta ramificación no se produce o si solo se realiza de forma incompleta, se crea un glucógeno que ya no se puede descomponer para un suministro rápido de energía. Por el contrario, se acumula muy rápidamente en el hígado, el bazo y los ganglios linfáticos. Después de cada comida, parte de la glucosa no utilizada se transporta al hígado para almacenarla como sustancia de reserva, el glucógeno.
Sin embargo, este material de reserva no se puede utilizar en su forma actual. La acumulación constante de glucógeno anormal agranda cada vez más el hígado y el bazo e inevitablemente conduce a la destrucción de ambos órganos.
Síntomas, dolencias y signos
La enfermedad de Andersen se manifiesta a través de una extraordinaria variabilidad. Se trata del almacenamiento constante de un glucógeno anormal, que ya no se puede descomponer. Pero la gravedad de la enfermedad puede ser diferente. Sin embargo, el pronóstico de la enfermedad de Andersen es en general muy malo. El síntoma más destacado es un hígado en constante crecimiento, a partir del cual se desarrolla rápidamente una cirrosis hepática.
La forma más grave se manifiesta a través de la falta o reducción de los movimientos del niño antes del nacimiento. El feto muestra signos de rigidez articular e hipoplasia pulmonar. Por lo general, en estos casos el niño nace muerto. En los casos clásicos, el niño todavía tiene un desarrollo normal al nacer. Sin embargo, en los primeros meses de vida se desarrollan hepatomegalia (agrandamiento del hígado) e hipotonía (falta de tensión muscular).
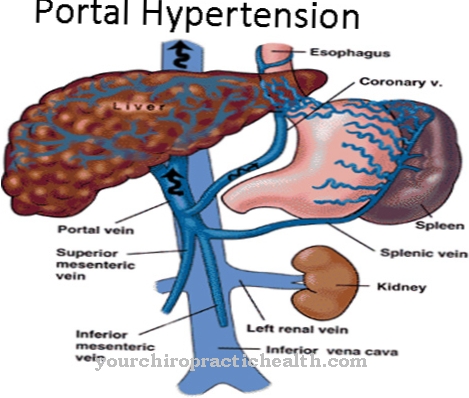
En general, el desarrollo del niño se retrasa. La enfermedad progresa rápidamente. El hígado desarrolla cirrosis. También hay un aumento de la presión portal y el bazo se agranda. Debido a la cirrosis del hígado, se desarrollan várices en el esófago con sangrado y ascitis correspondientes. La muerte suele ocurrir en la primera infancia. En casos más raros, la enfermedad comienza más tarde y muestra síntomas de debilidad muscular e insuficiencia cardíaca. Los síntomas neurológicos también ocurren aquí.
Diagnóstico y curso de la enfermedad
El diagnóstico se puede realizar en base al cuadro clínico y acompañado de pruebas de laboratorio, biopsias hepáticas y pruebas genéticas moleculares. En los exámenes histológicos, se nota la acumulación intracelular de estructuras similares a la amilopectina que se pueden teñir. La enzima responsable se examina en los hepatocitos, fibroblastos y leucocitos. Una deficiencia comprobada de amilo-1,4-1,6-transglucosidasa confirma el diagnóstico.
Complicaciones
Como regla general, la esperanza de vida del niño se reduce significativamente por la enfermedad de Andersen o el niño nace muerto. Esto puede provocar graves problemas psicológicos o depresión, especialmente con familiares o padres. En la mayoría de los casos, dependen de un tratamiento psicológico.
Los niños afectados padecen cirrosis del hígado, que finalmente conduce a la muerte. Además, las articulaciones también se ponen rígidas y los movimientos ya no son posibles debido a esta dolencia. El desarrollo mental del niño también se ve gravemente afectado por la enfermedad de Andersen, por lo que los afectados suelen depender siempre de la ayuda de otras personas. No es raro que ocurra insuficiencia cardíaca o debilidad muscular.
Los pacientes también pueden morir de muerte cardíaca. Desafortunadamente, la enfermedad de Andersen no se puede curar. El trasplante de hígado también solo puede aliviar los síntomas durante un período breve, ya que también se producirá el daño al hígado nuevo. Esto finalmente conduce a la muerte del niño. Hasta entonces, sin embargo, las quejas y los síntomas pueden limitarse con la ayuda de medidas médicas.
¿Cuándo deberías ir al médico?
La enfermedad de Andersen es una enfermedad genética que, en casos graves, puede provocar la muerte del feto en el útero. Por lo tanto, las mujeres embarazadas deben buscar tratamiento médico tan pronto como se noten irregularidades o anomalías durante el embarazo. Si la futura madre tiene la vaga sensación de que algo podría estar mal con el feto, debe consultar a un médico. Si el recién nacido sobrevive los primeros días y semanas después del nacimiento, se necesita un médico tan pronto como se hagan evidentes las peculiaridades en el curso posterior del desarrollo. Si tiene debilidad muscular o trastornos del movimiento, debe consultar a un médico.
Las alteraciones en el crecimiento son signos de una enfermedad existente y deben aclararse. Se deben examinar y tratar las anomalías cardíacas, las deformaciones del cuerpo y las discrepancias en el comportamiento del niño. En muchos casos, la enfermedad conduce a un agrandamiento de los órganos. El hígado o el bazo en particular se ven afectados en estos casos.
Por lo tanto, se necesita un médico tan pronto como se presente una forma inusual de la parte superior del cuerpo en comparación directa con bebés o niños de la misma edad. La decoloración de la piel u otras irregularidades en la apariencia de la piel son otros signos de deterioro de la salud. Un médico debe evaluar la cara o los ojos amarillentos.
Terapia y tratamiento
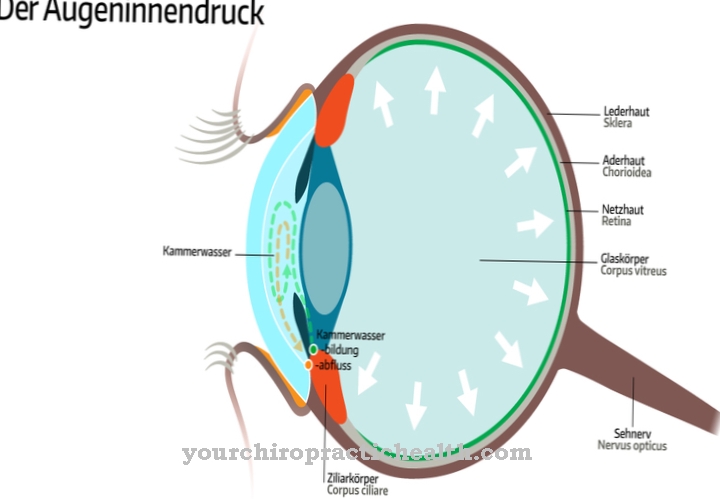
Dado que la enfermedad es genética, no se puede administrar un tratamiento causal. La terapia es solo sintomática. Como parte del tratamiento, los médicos se centran principalmente en las complicaciones que surgen. Esto reduce la presión en el circuito de la vena porta. También hay una sustitución de albúmina y factores de coagulación.
Si ocurre insuficiencia hepática, un trasplante de hígado puede prolongar la vida. Sin embargo, la enfermedad no se puede curar ni siquiera con un trasplante de hígado. El defecto genético está presente y también conducirá a depósitos de glucógeno anormal en el hígado nuevo. El almacenamiento del polisacárido defectuoso continúa en los otros órganos del llamado sistema reticulohistiocítico del bazo y los ganglios linfáticos, por lo que aún pueden ocurrir complicaciones graves incluso después de un trasplante de hígado exitoso.
El sistema reticulohistiocítico es parte del sistema inmunológico e incluye las células del tejido conectivo reticular. Estas células almacenan partículas y sustancias para descomponerlas y luego sacarlas del cuerpo. Sin embargo, la descomposición de las moléculas de polisacarosa defectuosas tampoco es posible aquí.
Outlook y pronóstico
La enfermedad de Andersen tiene un pronóstico relativamente malo. La enfermedad metabólica hasta ahora no ha sido curable y causa daño hepático severo. En algunos casos, se producen molestias musculares y enfermedades concomitantes que, si no se tratan, progresan progresivamente. La esperanza de vida está severamente limitada por la enfermedad. Los niños enfermos alcanzan una edad promedio de dos a cinco años. Un trasplante de hígado temprano mejora el pronóstico. El pronóstico es malo, especialmente para las formas clásicas de la enfermedad, especialmente si no hay trasplante de hígado en los primeros meses de vida.
Como regla general, el pronóstico a largo plazo se basa en la extensión, gravedad y progresión de la enfermedad. La enfermedad de Andersen es una de las glucogenosis más graves. La calidad de vida suele reducirse considerablemente debido a los problemas hepáticos y otros síntomas. Los analgésicos y la terapia integral mejoran el bienestar del niño, pero también están asociados con riesgos. El especialista en hígado responsable proporciona el pronóstico.
La esperanza de vida está severamente limitada por la enfermedad. Cualquier enfermedad concomitante que pueda ocurrir con enfermedades no detectadas también se incluye en el pronóstico. Por tanto, la enfermedad de Andersen ofrece un mal pronóstico en general. Los nuevos métodos de tratamiento podrían traer una mejora en el futuro.
prevención
Una prevención de la enfermedad de Andersen solo puede referirse al hecho de que la descendencia no hereda esta enfermedad. Dado que la enfermedad de Andersen se transmite de manera autosómica recesiva, se pueden omitir varias generaciones en la herencia. Si ya ha habido casos de enfermedad de Andersen en la familia y parientes, por lo tanto, se deben realizar pruebas genéticas en humanos.
Si el gen se encuentra en ambos padres, se recomienda el asesoramiento genético. En este caso, la descendencia tiene un 25 por ciento de posibilidades de desarrollar la enfermedad de Andersen.
Cura postoperatoria
Dado que la enfermedad de Andersen no se puede curar, el tratamiento de los síntomas y la contención de posibles complicaciones son el enfoque principal durante todo el período de tratamiento. La atención de seguimiento es necesaria después de las intervenciones que se llevan a cabo como parte de la terapia. Si se produce un trasplante de hígado, la atención de seguimiento profesional es muy importante para ello.
Después del procedimiento, esto asegura que el cuerpo no rechace el hígado nuevo. Los medicamentos especiales inhiben la respuesta inmunitaria del cuerpo. Sin embargo, como resultado, la resistencia del cuerpo a los patógenos se debilita, lo que debe tenerse en cuenta en la terapia posterior. Durante este tiempo, el paciente debe realizarse análisis de sangre periódicos. Se tiene cuidado para asegurarse de que no haya reacciones de rechazo u otras complicaciones graves, como disfunción renal, que pueden ocurrir como efectos secundarios.
Si bien los síntomas centrales de la enfermedad de Andersen se pueden mejorar directamente después de un trasplante de hígado, el depósito del glucógeno defectuoso continúa, por lo que se deben esperar complicaciones y síntomas progresivos incluso después de un trasplante. El especialista en hígado responsable puede proporcionar información más detallada sobre el pronóstico y el curso posterior del tratamiento.
Puedes hacerlo tu mismo
Las medidas de autoayuda que puede tomar un paciente con enfermedad de Andersen se limitan a inexistentes. Dado que la enfermedad tiene causas genéticas y no se puede controlar a pesar del tratamiento sintomático, las posibilidades de la persona afectada se agotan rápidamente. Es mejor que se tome en serio cualquier consejo sobre dieta y estilo de vida que le dé su médico tratante y que lo implemente.
Además, después de un trasplante de hígado, la persona afectada debe considerar un comportamiento suave. Se deben evitar el alcohol, los alimentos grasos y el esfuerzo. Esto hace que sea más fácil para el cuerpo aceptar realmente el nuevo órgano. Sin embargo, un trasplante exitoso, incluida la atención de seguimiento exitosa, no puede detener la glucogenosis tipo 4 en sí misma.
Dado que se trata de una enfermedad hereditaria autosómica recesiva (puede saltarse varias generaciones), tiene sentido elaborar un perfil genético al planificar una familia. Si bien los afectados por la enfermedad de Andersen ya conocen su gen, un análisis al respecto es particularmente valioso para los miembros de la familia. De esta manera, la transmisión del gen desencadenante se puede prevenir mediante una planificación familiar adecuada. Sin embargo, al menos se puede tener certeza sobre el riesgo de enfermedad en la propia descendencia.










.jpg)