Como Síndrome de Bourneville-Pringle Se conoce una tríada de tumores del cerebro con epilepsia y retraso en el desarrollo, lesiones cutáneas y crecimientos en otros sistemas orgánicos. La enfermedad es causada por una mutación de los dos genes TSC1 y TSC2. La terapia es sintomática con un enfoque en la epilepsia.
¿Qué es el síndrome de Bourneville-Pringle?

© Henrie - stock.adobe.com
El término médico síndrome de Bourneville-Pringle es sinónimo de esclerosis tuberosa. Este fenómeno patológico pertenece al grupo de enfermedades hereditarias y se caracteriza por tumores mayoritariamente benignos en la cara, en el cerebro y en el sistema de órganos, por discapacidad intelectual y ataques epilépticos.
La prevalencia de la esclerosis tuberosa en recién nacidos es de alrededor de un caso de cada 8.000 lactantes. Los neurólogos franceses Désiré-Magloire Bourneville y Édouard Brissaud describieron por primera vez la enfermedad junto con el dermatólogo británico John James Pringle en el siglo XIX. El nombre Síndrome de Bourneville-Pringle ha surgido por su bien.
En el mundo de habla inglesa, el complejo de síntomas se llama Complejo de esclerosis tuberosa. Clínicamente, el complejo se caracteriza por una tríada sintomática con los síntomas mencionados anteriormente. Una forma especial del síndrome es el síndrome del gen contiguo.
causas
Se han observado grupos familiares en relación con el síndrome de Bourneville-Pringle, aparentemente basados en una herencia autosómica dominante. Sin embargo, en la mitad de los casos, la enfermedad aparentemente se basa en una nueva mutación genética como causa. La tasa de mutaciones espontáneas es por lo menos tan alta como la de mutaciones heredadas.
En casos familiares, se observaron mutaciones en el gen TSC1 en el locus Chr.9q34 y en el gen TSC2 en el locus Chr.16p13 con la misma frecuencia. La aparición esporádica se limita casi exclusivamente a nuevas mutaciones en el gen TSC2. Ambos genes son genes supresores de tumores y, por tanto, participan en la supresión del crecimiento celular. Sus productos genéticos son la hamartina y la tuberina, cuyas funciones no se han aclarado de manera concluyente.
Las mutaciones en el contexto de un síndrome de Bourneville-Pringle se distribuyen por todos los exones de los genes mencionados y pueden corresponder a cualquier tipo de mutación. Todavía no se han observado grandes deleciones en el gen TSC2 en uno o más exones. La forma especial del síndrome del gen contiguo afecta tanto al gen TSC2 como al gen PKD1.
Síntomas, dolencias y signos
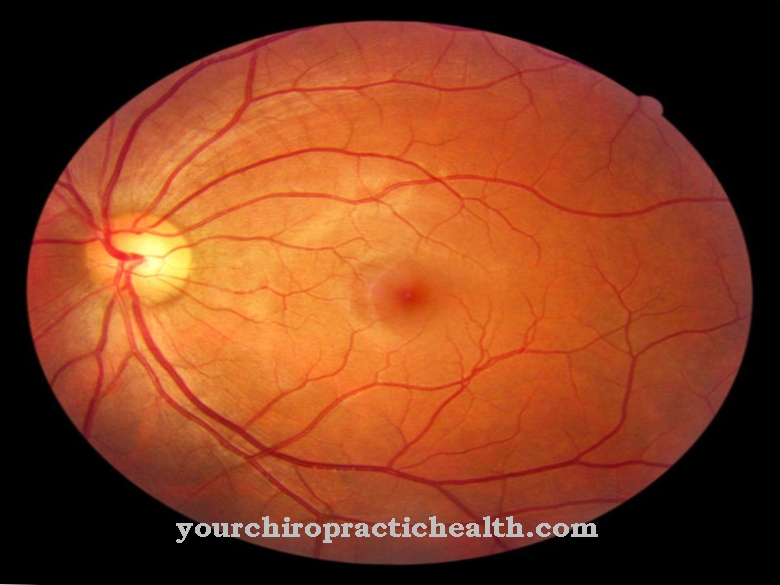
La esclerosis tuberosa se caracteriza por múltiples áreas de diferenciación tisular anormal, llamadas hamartias, que varían en ubicación con respecto a los sistemas de órganos. Los principales criterios de la enfermedad incluyen angiofibromas faciales y nevos de tejido conectivo en el área de la frente, angiofibromas no traumáticos, al menos tres manchas hipomelanóticas, nevos de tejido conectivo del sacro y varios hamartomas en la retina.
Además de la displasia cortical, también hay nódulos subependimarios, síntomas subependimarios de células gigantes y rabdomiomas del corazón. Además, las linfangiomiomatosis pulmonares y el angiomiolipoma de los riñones pueden mencionarse como criterios principales. Los síntomas que acompañan a los pacientes suelen presentar defectos en el esmalte dental, pólipos rectales o formaciones de quistes óseos.
Además, los síntomas pueden ir acompañados de una rigidez inusual de la sustancia blanca del cerebro. Lo mismo se aplica a los miomas gingivales, la despigmentación y los quistes de los riñones. La tríada del síndrome se divide en cambios sintomáticos de la piel, malformaciones del cerebro con trastornos del desarrollo y epilepsia y síntomas de otros sistemas orgánicos.
Diagnóstico y curso
Para diagnosticar la esclerosis tuberosa, el médico muestra al paciente dos criterios principales de la enfermedad o un síntoma principal con dos criterios secundarios. Los cambios en el cerebro generalmente se detectan antes y generalmente se muestran usando imágenes como una resonancia magnética. Un análisis genético molecular puede confirmar la sospecha de diagnóstico del síndrome y descartar síndromes similares a partir de un diagnóstico diferencial.
El pronóstico es bueno para los pacientes con síndrome de Bourneville-Pringle bastante leve. Muchos pacientes con TLP leve llevan una vida mayoritariamente normal. Las personas afectadas por DBP grave y, por tanto, epilepsia grave, deterioro cognitivo extremo y una gran cantidad de tumores tienen un pronóstico más precario y pueden tener que esperar efectos que acorten la vida.
Complicaciones
En el síndrome de Bourneville-Pringle o la esclerosis tuberosa, diferentes sistemas de órganos se ven afectados y pueden tener diferentes complicaciones. Por un lado, esta enfermedad afecta principalmente al sistema nervioso central y al cerebro. Los afectados sufren de epilepsia, especialmente en la infancia. Las convulsiones parciales son las más comunes, pero también pueden ser generalizadas.
Si no se trata, la epilepsia infantil puede convertirse en síndrome de Lennox-Gausaut. La persona afectada sufre una convulsión mayoritariamente tónica y ausencias varias veces al día, que en el peor de los casos puede convertirse en un estado epiléptico, una emergencia médica. A veces, también se pueden observar trastornos del desarrollo mental en el niño.
Además, un paciente puede desarrollar un aumento de la presión intracraneal durante el curso de la enfermedad. Esto conduce a fuertes dolores de cabeza y deterioro de la conciencia. En el peor de los casos, importantes centros de control pueden quedar atrapados en el área de la médula alargada (bulbo raquídeo), lo que puede provocar insuficiencia respiratoria.
La esclerosis tuberosa también puede ser la causa de quistes renales o tumores malignos, que pueden ser responsables de insuficiencia renal (insuficiencia renal). Esto limita gravemente la calidad de vida y es posible que el paciente deba someterse a diálisis o un trasplante. El rabdomioma intracardíaco puede desarrollarse en el corazón, que puede ser responsable de arritmias cardíacas o incluso muerte cardíaca.
¿Cuándo deberías ir al médico?
Si persisten las convulsiones epilépticas y el deterioro cognitivo, se debe consultar a un médico. Puede usar un examen de ultrasonido para determinar si el síndrome de Bourneville-Pringle es la causa. Un diagnóstico específico de la enfermedad tumoral solo es posible después de una anamnesis completa. La epilepsia característica ya se puede determinar en los primeros meses de vida.
Luego, el pediatra organizará un examen de rutina y diagnosticará rápidamente el síndrome de Bourneville-Pringle. Si los ataques epilépticos no ocurren, el diagnóstico es más difícil. Cualquier trastorno del desarrollo y problema de comportamiento a menudo solo se desarrolla en el transcurso de la niñez o la adolescencia. Básicamente: si el niño se comporta de forma anormal, tiene dificultades de aprendizaje o presenta otras deficiencias, se debe consultar al pediatra.
Otros signos de alerta que requieren aclaración médica son los cambios cutáneos crecientes como los chopos rojizos o las características manchas en forma de hoja en la piel. En el curso posterior, pueden aparecer tumores de piel, bultos y otras anomalías. Si uno o ambos padres tienen síndrome de Bourneville-Pringle, es aconsejable hacerse una evaluación médica durante el embarazo.
Doctores y terapeutas en su área
Tratamiento y Terapia
Hasta ahora, el síndrome de Bourneville-Pringle no se puede tratar de manera causal, ya que solo los enfoques de terapia génica pueden considerarse como terapia causal y estos enfoques son actualmente objeto de investigación, pero aún no se han aprobado para su uso. Por esta razón, actualmente solo se dispone de terapias sintomáticas para el tratamiento.
La terapia de la epilepsia es el foco de la terapia, ya que es precisamente este síntoma el que afecta gravemente la calidad de vida de los afectados y, en el peor de los casos, provoca un severo deterioro del estado de salud hasta la muerte. El tratamiento de la epilepsia es medicado o, en casos graves, quirúrgico en la medida de lo posible.
Por ejemplo, la separación de los dos hemisferios cerebrales mediante la extirpación quirúrgica del cuerpo calloso ha mostrado éxito en la terapia de la epilepsia en el pasado. Para formas más leves, la administración de antiepilépticos suele ser suficiente. Además de estos pasos de tratamiento, los tumores deben eliminarse de los sistemas de órganos. Como se trata en su mayoría de tumores benignos, no suele estar indicada la irradiación posterior.
Sin embargo, en el caso de un gran número de tumores, está indicada una estrecha vigilancia para identificar a tiempo posibles cambios que conduzcan a malignos. Dado que los afectados a menudo sufren de retraso mental, medidas como la intervención temprana también pueden ser pasos terapéuticos apropiados. El desarrollo del habla puede apoyarse con la logopedia.
Los retrasos en el desarrollo motor se pueden contrarrestar con medidas de fisioterapia y terapia ocupacional. Si la enfermedad genera estrés psicológico para el paciente, la psicoterapia también puede ser útil.
Outlook y pronóstico
Actualmente no existe una terapia curativa para el síndrome de Bourneville-Pringle. Solo es posible el tratamiento sintomático. La gravedad de la enfermedad es diferente para cada paciente. Por regla general, la esperanza de vida es normal. Sin embargo, esto puede reducirse con frecuentes ataques epilépticos, retraso mental severo y degeneración maligna de los tumores existentes.
La terapia se limita particularmente al tratamiento de ataques epilépticos. Como parte de la enfermedad, todos los tipos de convulsiones ocurren en la epilepsia. Se ha observado una asociación entre el desarrollo cognitivo y la frecuencia de las convulsiones. Los adultos experimentan principalmente convulsiones focales generalizadas secundarias.
En general, existen trastornos del desarrollo que se manifiestan en trastornos del lenguaje, el movimiento y el aprendizaje. El cociente de inteligencia del individuo afectado puede desarrollarse de manera diferente. Si bien esto es normal en la mitad de los pacientes, alrededor del 31 por ciento de los pacientes alcanzan un cociente de un máximo de 21.
Los cambios en la piel también son diferentes y dependen de la edad. Estos son adenomas de sebo. El tratamiento cosmético de los adenomas se realiza mediante extirpación quirúrgica o irradiación con láser. A menudo también existe un angiomiolipoma, un tumor benigno en el tejido renal. También se puede desarrollar un tumor benigno en los músculos estriados del corazón. Otros órganos, como los pulmones, también pueden verse afectados por tumores. La degeneración maligna es muy rara.
prevención
Hasta ahora, el síndrome de Bourneville-Pringle solo se puede prevenir si, en la planificación familiar, las parejas pueden usar pruebas genéticas moleculares para evaluar su riesgo de tener hijos enfermos y, si existe un mayor riesgo, deciden no tener sus propios hijos.
Puedes hacerlo tu mismo
La esclerosis tuberosa, también conocida como síndrome de Bourneville-Pringle, es una enfermedad genética que aún no puede tratarse de manera causal. Por tanto, las medidas terapéuticas comienzan con los síntomas.
La epilepsia es uno de los efectos secundarios más molestos que suele afectar gravemente a la calidad de vida de los afectados. Además del tratamiento farmacológico con fármacos antiepilépticos, los afectados a menudo pueden contribuir al hecho de que las convulsiones ocurren con menos frecuencia o son menos graves a lo largo de su estilo de vida. Los pacientes deben llevar un diario de la epilepsia para saber si los factores de su vida diaria están desencadenando las convulsiones.
Tales factores pueden ser de naturaleza completamente diferente. Ciertos alimentos, alcohol, drogas que alteran la mente, así como falta de sueño, estrés, sentimientos severos de ansiedad o, en las mujeres, períodos menstruales. Los factores críticos deben evitarse en la medida de lo posible. Muchos enfermos y sus familiares también se benefician de unirse a un grupo de autoayuda para epilépticos, que ahora existe en numerosas ciudades alemanas.
Muy a menudo, los afectados por la esclerosis tuberosa también sufren de retraso en el desarrollo intelectual. Las consecuencias negativas pueden contrarrestarse mediante una intervención temprana adecuada. Los padres pueden obtener consejos de los pediatras o de la oficina de bienestar juvenil. Si el desarrollo de las habilidades motoras también se ve afectado, las medidas ocupacionales y fisioterapéuticas ayudan. En el caso de retraso en el desarrollo del lenguaje, se debe consultar a un logopeda.

.jpg)









.jpg)