En el Síndrome de Saethre-Chotzen es una enfermedad asociada con craneosinostosis. El síndrome de Saethre-Chotzen es congénito ya que las causas son de naturaleza genética. La enfermedad se conoce con la abreviatura SCS. Los principales síntomas del síndrome de Saethre-Chotzen son sinostosis de la sutura coronaria en uno o ambos lados, ptosis, cara asimétrica, orejas inusualmente pequeñas y estrabismo.
¿Qué es el síndrome de Saethre-Chotzen?

© Matthieu - stock.adobe.com
los Síndrome de Saethre-Chotzen ocurre relativamente raramente. Con una frecuencia estimada de 1: 25.000 a 1: 50.000, el síndrome de Saethre-Chotzen se presenta con una frecuencia algo más frecuente que muchas otras enfermedades hereditarias. Las malformaciones craneofaciales son típicas del síndrome de Saethre-Chotzen. Una craneosinostosis se produce junto con un sinfalangismo y una sindactilia.
Algunos médicos se refieren al síndrome de Saethre-Chotzen con los términos sinónimos Síndrome de Chotzen o Síndrome de acrocefalosindactilia tipo III designado. Básicamente, el nombre común de síndrome de Saethre-Chotzen se remonta a aquellas personas que describieron científicamente la enfermedad por primera vez en 1931. Estos son los dos médicos Chotzen y Saethre.
Además, existe una forma especial de síndrome de Saethre-Chotzen, en la que los pacientes también presentan anomalías en los párpados además de los síntomas típicos. Esta peculiaridad se conoce con el término síndrome de Robinow-Sorauf. Como parte de esta enfermedad, los dedos gordos del pie del paciente están partidos o, a veces, se doblan. Hasta ahora se ha asumido que el síndrome de Robinow-Sorauf es una variación alélica del síndrome de Saethre-Chotzen.
causas
Los factores responsables de la patogénesis del síndrome de Saethre-Chotzen son defectos genéticos. Con respecto al desarrollo del síndrome de Saethre-Chotzen, tanto las deleciones como las mutaciones puntuales son particularmente relevantes aquí. Estos procesos defectuosos tienen lugar en el llamado gen TWIST1. Un factor de transcripción especial se codifica en el locus correspondiente. Esto define líneas de células y participa en la diferenciación.
Debido a los defectos genéticos de este gen, las suturas del cráneo se fusionan demasiado pronto. Las deleciones dan como resultado una apariencia de los pacientes afectados que se desvía significativamente de la media. Además, en algunos casos son responsables del hecho de que algunas personas con síndrome de Saethre-Chotzen padecen de capacidades mentales deterioradas.
Síntomas, dolencias y signos
Los síntomas clínicos del síndrome de Saethre-Chotzen varían de un caso a otro. Normalmente, como recién nacidos, las personas afectadas tienen una sinostosis que surge en el área de la sutura coronaria. Esta costura también se llama costura lambda o costura de flecha.
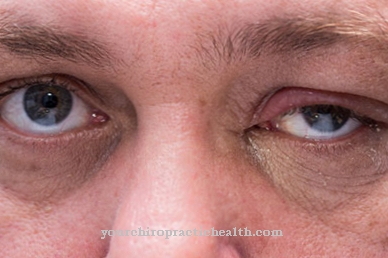
Debido a esto, el cráneo del paciente tiene una forma inusual. Además, suele haber claras asimetrías en el rostro de la persona. Además, los pacientes suelen tener una frente muy alta con una línea de implantación profunda, orejas pequeñas y estrabismo. También son frecuentes la ptosis, braquidactilia y estenosis de los conductos lagrimales.
Los dedos medio e índice a menudo están parcialmente conectados entre sí en el área de la piel. En la mayoría de los casos, las personas con síndrome de Saethre-Chotzen tienen un cociente intelectual medio. Sin embargo, algunos de los afectados muestran retrasos importantes en el desarrollo intelectual.
Algunas de las personas con síndrome de Saethre-Chotzen desarrollan pérdida auditiva durante la niñez. Suele ser una pérdida auditiva conductiva. En casos raros, además de los síntomas generales, los pacientes padecen baja estatura, maxilar superior hipoplásico, deformidades del corazón o hipertelorismo.
Además, algunos de los pacientes presentan paladar hendido, malformaciones de los huesos vertebrales, úvula bífida y apnea obstructiva del sueño. Si la sinostosis es particularmente pronunciada, el paciente afectado puede experimentar problemas de visión, dolor en el área de la cabeza debido al aumento de la presión interna en el cráneo y ataques epilépticos. En casos extremos, las personas mueren como resultado de sus síntomas.
Diagnóstico y curso de la enfermedad
El diagnóstico del síndrome de Saethre-Chotzen generalmente lo realiza un médico especialista. El médico de cabecera o el pediatra derivan al paciente al especialista correspondiente. Una primera anamnesis en presencia del custodio de los pacientes, en su mayoría niños, proporciona información sobre los síntomas actuales.
El examen clínico se basa en los síntomas típicos, que en algunos casos ya indican claramente el síndrome de Saethre-Chotzen. La atención se centra en los exámenes visuales del fenotipo y varios métodos de imagen. El médico tratante utiliza, por ejemplo, técnicas de rayos X y TC. Suelen centrarse en la zona del cráneo, las extremidades y la columna.
En última instancia, es posible realizar un diagnóstico definitivo del síndrome de Saethre-Chotzen con la ayuda de una prueba genética de laboratorio. Si se identifica una deleción o una mutación puntual en el gen responsable, el diagnóstico de síndrome de Saethre-Chotzen se considera confirmado. El diagnóstico diferencial también juega un papel importante, ya que algunos síntomas del síndrome de Saethre-Chotzen a veces se confunden. El médico descarta el síndrome de Crouzon, el síndrome de Baller-Gerold, el síndrome de Pfeiffer y el síndrome de Muenke.
Complicaciones
Pueden ocurrir varias complicaciones como resultado del síndrome de Saethre-Chotzen. Los afectados suelen sufrir retrasos importantes en el desarrollo intelectual. Esto puede conducir a la exclusión social y, como resultado, al desarrollo de complicaciones psicológicas como depresión o complejos de inferioridad. Como resultado de la pérdida auditiva, puede haber más restricciones en la vida diaria y la calidad de vida de los afectados se reduce.
Si hay deformidades del corazón, puede ocurrir insuficiencia cardíaca como consecuencia a largo plazo. Además, en el curso de la enfermedad, los ataques epilépticos ocurren repetidamente, lo que aumenta el riesgo de accidentes. En el peor de los casos, los afectados mueren por las consecuencias de las distintas quejas. Incluso con un resultado positivo, inevitablemente surgen complicaciones físicas y mentales. El tratamiento quirúrgico de las malformaciones individuales siempre está asociado con riesgos.
Durante una operación pueden producirse hemorragias e infecciones, así como lesiones nerviosas. Esto puede provocar trastornos en la cicatrización de heridas, inflamación y cicatrices. Los medicamentos prescritos para acompañar el tratamiento quirúrgico deben adaptarse con precisión a la condición del paciente. De lo contrario, pueden producirse efectos secundarios e interacciones potencialmente mortales debido a los diversos trastornos mentales y físicos.
¿Cuándo deberías ir al médico?
El síndrome de Saethre-Chotzen siempre debe ser examinado y tratado por un médico. Si no se trata, puede ocasionar complicaciones graves que limitan gravemente la vida de la persona afectada. Dado que este síndrome es un trastorno genético, tampoco se puede curar por completo. Por tanto, el paciente depende de un tratamiento puramente sintomático. Si el interesado desea tener hijos, también se puede realizar un asesoramiento genético para evitar que el síndrome se transmita a los hijos.
Se debe consultar a un médico si el paciente sufre un retraso significativo en el desarrollo. Sobre todo, el desarrollo intelectual está severamente restringido. Una pérdida de audición o una baja estatura también pueden indicar el síndrome de Saethre-Chotzen y siempre deben ser examinados por un médico. En la mayoría de los casos, también se producen deformidades del corazón, por lo que la persona afectada depende de exámenes cardíacos periódicos.
Un médico de cabecera o un pediatra pueden realizar un examen inicial y un diagnóstico del síndrome de Saethre-Chotzen. Sin embargo, el tratamiento adicional lo llevan a cabo varios especialistas y depende de la gravedad de las malformaciones.
Tratamiento y Terapia
Los pacientes con síndrome de Saethre-Chotzen ya se someten a diversas intervenciones quirúrgicas en la infancia. Generalmente se utiliza una craneoplastia. Las vías respiratorias estrechas se ensanchan y el paladar hendido se cierra. Las revisiones periódicas controlan el desarrollo auditivo, intelectual y motor. También es importante reconocer a tiempo la posible ambliopía. Lo mismo se aplica al edema papilar con curso crónico.
prevención
Según el estado actual de la investigación, el síndrome de Saethre-Chotzen no se puede prevenir.
Cura postoperatoria
El alcance de las medidas de seguimiento para el síndrome de Saethre-Chotzen (SCS) se basa en la naturaleza de los síntomas de la enfermedad y su respectiva gravedad. Sintomáticamente, el SCS puede causar deformaciones de la cabeza, la zona media de la cara, la columna, las extremidades y las articulaciones. En la mayoría de los casos de enfermedad, las partes del cuerpo antes mencionadas no se desarrollan correctamente. Enfermedades secundarias graves pueden seguir a la deformación.
La tarea de la atención de seguimiento es identificar y tratar las enfermedades secundarias del SCS lo antes posible. El foco de las medidas de seguimiento es la observación del curso de la enfermedad. Las deformaciones en el área de la cabeza, por ejemplo, generalmente se remodelan quirúrgicamente. De este modo se puede evitar la ceguera de la persona afectada. Durante la atención de seguimiento de la remodelación, un oftalmólogo examina los nervios ópticos de la persona afectada cada trimestre hasta los 12 años.
Los exámenes de seguimiento tienen como objetivo identificar lo antes posible si el cráneo es demasiado estrecho y, si corresponde, existe riesgo de ceguera y en qué medida. Con la remodelación del cráneo, pueden ocurrir ataques epilépticos en casos individuales. Estos pueden tratarse con medicamentos durante el cuidado posterior.
A medida que avanza la enfermedad, el desarrollo de la epilepsia se puede controlar de cerca neurológicamente mediante registros electroencefalográficos (EEG). Las deformaciones en las cuatro partes restantes del cuerpo conducen regularmente a una movilidad restringida sintomática. El postratamiento se centra básicamente en aumentar la movilidad mediante fisioterapia. Las partes doloridas del cuerpo se tratan con medicamentos.
Puedes hacerlo tu mismo
Las posibilidades de autoayuda en el síndrome de Saethre-Chotzen están orientadas al manejo óptimo de la enfermedad. El paciente y sus familiares están expuestos a varios desafíos de afrontamiento, para los cuales deben estar preparados lo más ampliamente posible.
La autoconfianza del paciente debe construirse en una etapa temprana. Debido a las anomalías visuales, la alteración representa una dificultad particular en la vida cotidiana, que el paciente debe poder afrontar emocionalmente. La ayuda y el apoyo de un psicoterapeuta también se pueden utilizar para esto. Dado que las habilidades mentales están restringidas, se debe prestar atención al desarrollo del niño. No se permite una comparación con compañeros de juegos de la misma edad. El proceso de aprendizaje también debe adaptarse a las capacidades y habilidades del paciente.
En el curso de la enfermedad, a menudo hay varias intervenciones quirúrgicas en los primeros años de vida. Para hacer frente a estos desafíos, el organismo debe recibir el apoyo de una dieta saludable rica en vitaminas. La higiene óptima del sueño y evitar el estrés también ayudan a aliviar los síntomas. Además de las malformaciones, muchos pacientes también sufren pérdida de audición. Este hecho debe tenerse en cuenta en la vida cotidiana a la hora de reducir el riesgo de accidentes y lenguaje. La formación dirigida ayuda a mejorar las habilidades lingüísticas y la comunicación con otras personas.

.jpg)




.jpg)

.jpg)