El médico al que se refiere como Síndrome de hiper-IgD - también conocida como HIDS - una enfermedad hereditaria que se caracteriza predominantemente por ataques recurrentes de fiebre. Por esta razón, el síndrome de hiper-IgD también es uno de los llamados síndromes de fiebre periódica.
Durante los episodios febriles, los afectados se quejan de diarrea, dolor abdominal y también náuseas y vómitos. No existe un tratamiento causal; sin embargo, el pronóstico, en términos de esperanza de vida del paciente, es bueno.
¿Qué es el síndrome de hiper-IgD?

© izholudeva - stock.adobe.com
los Síndrome de hiper-IgD (o HIDS) es una enfermedad hereditaria. Son característicos los ataques recurrentes de fiebre y síntomas gastrointestinales. Las enfermedades periódicas, a las que pertenece el síndrome de hiper-IgD, se conocen desde el siglo XIX, pero no fueron redefinidas y moldeadas hasta 1948 por el médico Hobart A. Reimann.
En 1984 se mencionó por primera vez el síndrome de hiper-IgD. Un grupo de trabajo holandés describió fiebre recurrente en hermanos, así como reacciones inflamatorias generales, así como un aumento significativo de inmunoglobulina-D e inmunoglobulina-A. Se han realizado alrededor de 160 diagnósticos desde 2001; la mayoría de los afectados vive en Francia y los Países Bajos. Sin embargo, se cree que el número de casos no denunciados es mayor.
causas
El síndrome de hiper-IgD surge debido a un cambio en la información genética, lo que se conoce como mutación. La mutación tiene lugar en el cromosoma 12. La herencia del síndrome de hiper-IgD es autosómica recesiva. En alrededor del 80 por ciento de todos los casos, hay principalmente una mutación sin sentido en el área del gen, que posteriormente codifica la MVK (12q12, GeneID 4598 - enzima mevalonato quinasa). Esta mutación provoca una estabilidad ligeramente reducida de la actividad de la enzima.
Sin embargo, hasta ahora no está claro por qué la actividad reducida de la mevalonato quinasa desencadena posteriormente brotes. Debido a la causa poco clara, tampoco existe una terapia conocida que a veces combata la causa. Por lo tanto, los médicos solo pueden realizar tratamientos sintomáticos que se centren principalmente en los ataques de fiebre.
Síntomas, dolencias y signos
El síndrome de hiper-IgD se presenta desde el primer año de vida. Son característicos los ataques recurrentes de fiebre; el paciente se queja de fiebre y escalofríos que se desarrollan rápidamente. Los desencadenantes de esos ataques de fiebre son lesiones menores, vacunas o incluso estrés y operaciones.
En muchos casos, los pacientes también se quejan de dolor abdominal, náuseas y vómitos y diarrea, entre otras cosas. A veces, los ganglios linfáticos cervicales también pueden hincharse. Otros síntomas que son característicos del síndrome de hiper-IgD: dolor de cabeza, inflamación de las articulaciones así como dolor y erupciones en las articulaciones.
Las recaídas en sí ocurren a intervalos de entre cuatro y seis semanas. La duración es de entre tres y siete días. Sin embargo, la duración y la frecuencia de las recaídas pueden variar significativamente según el paciente. Los ataques de fiebre ocurren con mayor frecuencia, especialmente en la niñez; en la edad adulta la frecuencia e intensidad disminuyen.
Diagnóstico y curso de la enfermedad
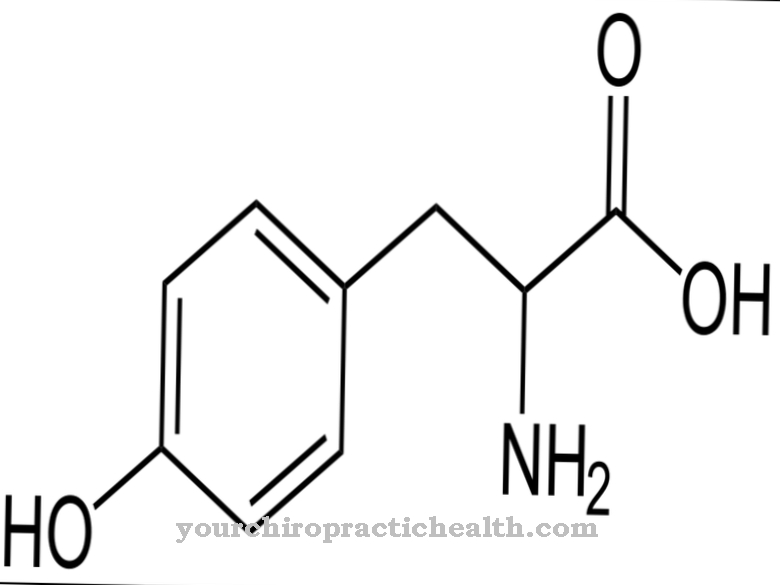
Si hay síntomas característicos que sugieran síndrome de hiper-IgD, verificar la concentración de inmunoglobulina D en la sangre puede proporcionar información sobre si el síndrome de hiper-IgD realmente existe. Si la concentración de inmunoglobulina D es superior a 100 UI / ml, se puede suponer que existe un síndrome de hiper-IgD. La evidencia genética molecular de una mutación existente a veces también puede proporcionar información sobre si hay o no un síndrome de hiper-IgD.
Sin embargo, para asegurar el diagnóstico, el médico debe descartar previamente otras enfermedades con síntomas similares. El síndrome de hiper-Ig-D es también uno de los síndromes febriles periódicos; También se debe descartar previamente la fiebre mediterránea familiar (FMF).
Otras enfermedades que tienen un curso similar y deben descartarse antes del diagnóstico de síndrome de hiper-IgD son: el "síndrome periódico asociado al receptor del factor de necrosis tumoral 1" (también conocido como TRAPS), la neutropenia cíclica y las enfermedades crónicas-infantiles-neurológicas. -síndrome articular cutáneo (o síndrome CINCA) y síndrome de Muckle-Wells. También incluye enfermedades que caen bajo el síndrome PFAPA (fiebre periódica, aftas, síndrome de adenitis o faringitis).
Incluso si el tratamiento del síndrome de hiper-IgD es relativamente difícil, el pronóstico sigue siendo bueno. La esperanza de vida no está limitada, incluso si se trata de una forma extremadamente grave del síndrome. Sin embargo, las articulaciones pueden ser atacadas en el curso de los ataques, por lo que es posible la destrucción articular.
La amiloidosis, como en la fiebre mediterránea familiar, hasta ahora solo se ha documentado en casos aislados. En algunos casos, también se documentaron anomalías neurológicas y limitaciones en las capacidades intelectuales. Los trastornos de la epilepsia, la coordinación y el equilibrio también son posibles, pero también ocurren raramente.
Complicaciones
Debido al síndrome de hiper-IgD, la persona afectada sufre ataques intensos de fiebre, que se repiten después de cierto tiempo. No es posible predecir la hora exacta del próximo ataque de fiebre. Además de la fiebre, la persona afectada también sufre de dolor abdominal y diarrea. Además, también hay náuseas y vómitos.
La calidad de vida se ve muy reducida por el síndrome de hiper-IgD y los síntomas dificultan la vida cotidiana. Como regla general, la capacidad de recuperación del paciente también se reduce y la persona afectada parece agotada y cansada. Los ataques de fiebre también pueden tener un efecto negativo en la psique y provocar depresión u otros trastornos.
También hay dolor de cabeza y articulaciones. No es raro que la piel se vea afectada por erupciones y los ganglios linfáticos se inflamen. Como regla general, los niños en particular se ven afectados por los frecuentes ataques de fiebre. El tratamiento del síndrome de hiper-IgD es sintomático y limita los síntomas del ataque febril. No hay más complicaciones. En la mayoría de los casos, la frecuencia disminuye con la edad. La esperanza de vida tampoco se ve reducida por el síndrome de hiper-IgD.
¿Cuándo deberías ir al médico?
En el caso del síndrome de hiper-IgD, los síntomas graves aparecen repentinamente después del primer año de vida por lo que siempre es necesaria la consulta de un médico. Dado que también se observan síntomas similares en otras enfermedades, es necesaria una aclaración diagnóstica urgente de los síntomas. Ésta es la única forma de iniciar una terapia eficaz. Se debe realizar una visita al médico, entre otras cosas, si el niño de un año sufre repentinamente ataques inexplicables de fiebre, que pueden ocurrir a intervalos más prolongados.
La fiebre suele anunciarse con la aparición de escalofríos. Sobre todo, también se debe consultar al médico si hay un ataque de fiebre en ocasiones especiales como vacunas, estrés o lesiones. Los síntomas que acompañan a la fiebre ya deberían utilizarse como una oportunidad para buscar ayuda médica. Estos síntomas incluyen inflamación de los ganglios linfáticos, dolor abdominal intenso, vómitos y diarrea.
Si el médico diagnostica el síndrome de hiper-IgD, discutirá la terapia necesaria con los padres. Consiste en la administración del agente reductor del colesterol simvastatina y el inmunosupresor etanercept. Con estos fármacos se alivian los síntomas y al mismo tiempo se reduce el riesgo de complicaciones raras como contracturas articulares o adherencias en el abdomen. Dado que la enfermedad no suele afectar la esperanza de vida y los ataques de fiebre son cada vez menos frecuentes a lo largo de la vida, no es necesaria una atención médica constante.
Doctores y terapeutas en su área
Terapia y tratamiento
Hasta el momento no existe una terapia o tratamiento que combata o trate principalmente la causa del síndrome de hiper-IgD. Por esta razón, los médicos se concentran principalmente en el tratamiento sintomático de los ataques de fiebre recurrentes. Sin embargo, estos tratamientos también resultan extremadamente difíciles, ya que no solo difieren de un paciente a otro, sino que también pueden tener diferentes intensidades según el episodio.
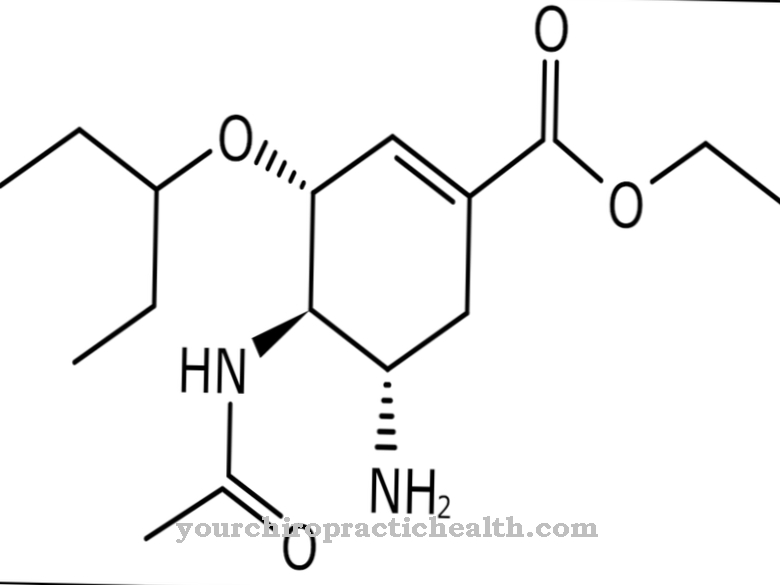
Los fármacos antipiréticos o antiinflamatorios clásicos, como los antiinflamatorios no esteroides, son completamente ineficaces. La colchicina, los esteroides o la talidomida también han demostrado ser ineficaces.Sin embargo, los profesionales médicos han reconocido que la simvastatina puede ayudar a tratar cualquier brote.
Hace algún tiempo, también se informaron avances positivos en el contexto del "anakinra análogo de interleucina-1ra". En ocasiones, el “etanercept, antagonista del factor de necrosis tumoral α” puede reducir los días de fiebre y aliviar la intensidad. Actualmente no se conocen otros tratamientos.
Puedes encontrar tu medicación aquí
➔ Medicamentos para la fiebre y los escalofríosOutlook y pronóstico
El síndrome de hiper-IgD ofrece un pronóstico relativamente positivo. Aunque la enfermedad se asocia con síntomas graves como molestias gastrointestinales, erupciones cutáneas, daño en las articulaciones y fiebre, a largo plazo estos problemas de salud pueden reducirse en gran medida o incluso eliminarse por completo con un tratamiento farmacológico integral.
Incluso si las articulaciones están afectadas, la enfermedad no siempre se asocia con síntomas a largo plazo, siempre que se utilice un tratamiento temprano. Solo en casos aislados se producen trastornos articulares permanentes, que deterioran permanentemente la calidad de vida y el bienestar y, por tanto, conllevan el riesgo de sufrimiento mental. La amiloidosis, que puede afectar a todos los órganos y al sistema endocrino, solo ocurre en unos pocos pacientes. Sin embargo, algunos pacientes experimentan trastornos neurológicos que pueden afectar las capacidades mentales y la coordinación. En casos graves, se puede desarrollar epilepsia.
En la mayoría de los casos, los afectados pueden llevar una vida sin dolor, lo que, sin embargo, siempre se asocia con tratamientos farmacológicos a largo plazo y medidas fisioterapéuticas. Las personas enfermas también tienen que someterse a intervenciones quirúrgicas una y otra vez o sufrir efectos secundarios e interacciones como resultado de la administración constante de medicamentos. Los problemas de salud mental también pueden ocurrir en pacientes con HIDS con enfermedades crónicas. Las complicaciones psicológicas típicas son los complejos de inferioridad y los estados de ánimo depresivos hasta la depresión severa. La esperanza de vida no se reduce por el síndrome de hiper-IgD.
prevención
Debido al hecho de que el síndrome de Hiper-IgD es una enfermedad hereditaria, las medidas preventivas no se conocen ni son posibles.
Cura postoperatoria
Las medidas u opciones para la atención de seguimiento suelen ser muy limitadas en el caso del síndrome de hiper-IgD. Dado que también es una enfermedad hereditaria, no se puede lograr una curación completa. Por lo tanto, los afectados dependen de una terapia y un tratamiento de por vida para aliviar los síntomas de forma permanente.
Para evitar transmitir el síndrome de Hiper-IgD a los descendientes, también se debe brindar asesoramiento genético si el niño desea tener hijos. Ésta es la única forma de prevenir la transmisión de la enfermedad. Dado que el síndrome de hiper-IgD suele tratarse con medicación, los afectados deben asegurarse de que se tomen de forma regular y, sobre todo, de forma correcta.
También se deben tener en cuenta las posibles interacciones. Si tiene alguna pregunta o no está clara, siempre debe comunicarse primero con un médico. Dado que el síndrome de hiper-IgD también puede promover tumores, los afectados deben ser examinados periódicamente por un médico. El apoyo y el cuidado de amigos y familiares también es muy importante y puede aliviar los síntomas. Sobre todo, se previenen las quejas psicológicas o la depresión. La esperanza de vida de los afectados puede verse reducida por el síndrome de hiper-IgD.
Puedes hacerlo tu mismo
Dado que, lamentablemente, el síndrome de hiper-IgD no puede tratarse de manera causal y, por lo tanto, causal, solo se pueden restringir los síntomas y las quejas individuales.
Los ataques de fiebre se tratan con medicamentos. El reposo en cama general y el cuidado de su propio cuerpo también pueden tener un efecto muy positivo en el curso de la enfermedad. Sobre todo, los afectados no deben simplemente realizar actividades físicas. El uso de medicamentos antipiréticos o analgésicos también puede tener un efecto negativo en el estómago, por lo que se debe consultar a un médico.
Además, el contacto con otras personas afectadas también puede ser útil en el caso del síndrome de hiper-IgD. Esto puede hacer que la vida diaria sea más fácil de afrontar. La ayuda de amigos y conocidos también puede reducir y aliviar significativamente los síntomas psicológicos del síndrome. Desafortunadamente, los afectados tienen que aceptar la enfermedad y no deben olvidar que los ataques de fiebre ocurren solo por un corto período de tiempo. Sin embargo, el tratamiento sigue siendo necesario para las convulsiones epilépticas. En particular, la conducción de vehículos o el manejo de maquinaria pesada no debe realizarse en casos de epilepsia.
.jpg)

.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)