Bajo el término Ciliopatía Se resumen diferentes enfermedades genéticas, que provocan un mal funcionamiento de los cilios o de las células que los portan.
¿Qué es la ciliopatía?
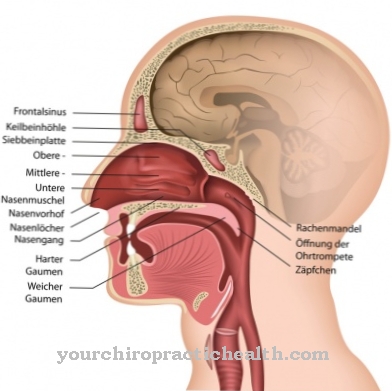
Los cilios (también llamados cilios de cine) son protuberancias citoplásmicas de la célula. Tienen hasta 10 µm de largo y hasta 0,25 µm. Los cilios pueden moverse libremente y se utilizan para transportar películas líquidas y mucosas. Los cilios del cine se golpean uniformemente uno detrás del otro, lo que conduce a una corriente de parpadeo uniforme.
Esto asegura que el líquido o la mucosidad se eliminen. En las diversas enfermedades que se llaman Ciliopatías se combinan, los cilios o las células que los portan se dañan, lo que significa que ya no pueden realizar sus tareas de manera suficiente.
causas
Las ciliopatías son enfermedades genéticas. Ahora se puede asignar un gran número de enfermedades hereditarias a las ciliopatías; para algunos, la prueba final aún no se ha proporcionado. Entonces, la ciliopatía es heredable.
Las ciliopatías individuales difieren considerablemente entre sí, ya que los cilios se encuentran en muchas partes diferentes del cuerpo humano y realizan diferentes tareas allí. Como resultado, los síntomas y el diagnóstico de las ciliopatías no son uniformes, pero hay una diferencia considerable entre las enfermedades individuales.
Síntomas, dolencias y signos
Los síntomas, quejas y signos de la ciliopatía no son uniformes, ya que la ciliopatía no es un cuadro clínico cerrado, sino simplemente un término general para varias enfermedades hereditarias de los cilios. Por esta razón, no es posible un diagnóstico estandarizado. Las ciliopatías incluyen una amplia variedad de enfermedades con síntomas a veces muy diferentes:
- Síndrome de kartagener
- Síndrome de Meckel-Gruber
- Síndrome de Joubert
- Síndrome de Laurence Moon Biedl Bardet (LMBBS)
- Nefronoptisis
- Síndrome de Løken mayor
- Quiste de hígado
- Síndrome de Bardet-Biedel
- Síndrome de Ellis van Creveld (EVC)
- algunos tipos de retinopatía
- ciertas formas de hidrocefalia
- Síndrome oro-facio-digital tipo 1 (OFD)
- Riñones quísticos hereditarios recesivos y dominantes (ARPKD, ADPKD)
- Enfermedad renal quística medular (ADMCKD)
- Síndrome de Alström (ALMS)
- Síndrome de polidactilia de costilla corta (SRPS)
- Displasia craneoectodérmica
- displasia torácica asfixiante (DAT)
A continuación, se presentan como ejemplos los síntomas y el diagnóstico del síndrome de Kartagener y el síndrome de Laurence-Moon-Biedl-Bardet (LMBBS).
El síndrome de Kartagener, también conocido como discinesia ciliar primaria, es una cilioptia. En este síndrome existe una disfunción de las células ciliadas, especialmente del epitelio ciliado respiratorio. El movimiento de los cilios se ve alterado, por lo que las secreciones no pueden o no pueden eliminarse lo suficiente.
De ello se deduce que el mecanismo de autolimpieza de los bronquios (aclaramiento mucociliar), que es transportado por el epitelio ciliado respiratorio, está gravemente afectado en este síndrome, que se debe a la disfunción de los cilios. El síndrome de Kartagener generalmente se hereda como un rasgo autosómico recesivo.
Todas las células del cuerpo ocupadas por cilios se ven afectadas, es decir, además del epitelio ciliado respiratorio, también se ven afectadas las células de la trompeta del oído (tubo auditivo) y los senos paranasales. El foco de los síntomas, sin embargo, está en los bronquios, lo que se debe al hecho de que la mayoría de las células ocupadas por cilios se encuentran allí.
En aproximadamente el 50 por ciento de los pacientes afectados por el síndrome de Katagener, se produce una posición anormal de los órganos internos durante la fase embrionaria en forma de situs invertus, una disposición en espejo de los órganos y vasos. Los hombres afectados son en su mayoría estériles debido a la discinesia alterada de los espermatozoides. La mayoría de las mujeres son estériles debido a la discinesia de los cilios alrededor de las trompas de Falopio.
La mayoría de los síntomas ocurren en el tracto respiratorio. En el curso de la autolimpieza alterada de los bronquios, a menudo se producen obstrucciones e infecciones de los bronquios. Las bronquitis son en su mayoría recurrentes y difíciles de tratar. Además, existen frecuentes rinitis, sinusitis y otitis media recurrentes y poco tratables.
En el curso de la enfermedad, a menudo se producen bronquiectasias, una expansión irreversible de los bronquios. El síndrome de dificultad respiratoria puede ocurrir en recién nacidos. La hidrocefalia, una expansión de los espacios de licor internos o externos, también puede ocurrir en los recién nacidos.
El síndrome de Kartagener a menudo se nota por una mayor susceptibilidad a las infecciones en la infancia. Si hay un situs invertus además de una mayor susceptibilidad a la infección, se puede suponer la existencia del síndrome de Kartagener. Si falta el situs invertus, el diagnóstico es difícil. La evidencia de la presencia del síndrome de Kartagener se obtiene mediante un examen microscópico electrónico de frotis con cepillo o biopsias de las membranas mucosas relevantes.
No es posible una terapia causal. El tratamiento es sintomático. Con un diagnóstico temprano y una terapia adecuada, los afectados pueden llevar una vida relativamente normal.
El síndrome de Laurence-Moon-Bardet-Biedl también es una de las ciliopatías. Se caracteriza por una amplia variedad de mutaciones y malformaciones que se desencadenan por mutaciones en diferentes cromosomas o ubicaciones de genes. La herencia es autosómica recesiva. Pueden ocurrir diversos síntomas según los genes afectados.
Estos no son igualmente pronunciados en todos los pacientes. Estos síntomas incluyen: obesidad; hipertensión arterial; Diabetes mellitus; Baja estatura; Hipotensión muscular; Malformaciones del hígado, ovarios y vías biliares; Hipogonadismo; Hipoplasia renal; Insuficiencia renal; Pielonefritis; trastornos motores; Discapacidad mental; Retinitis pigmentosa; Ceguera; Anosmia; Pérdida de la audición; Hemeralopía; un cuello corto; esquinas notables del párpado; Polidactilia y sindactilia.
El diagnóstico de LMBBS es difícil porque los síntomas también ocurren en una variedad de otras enfermedades. Al igual que con el síndrome de Kartagener, el diagnóstico final se realiza mediante una prueba de biología molecular. No existe una terapia causal; el tratamiento es sintomático.
Diagnóstico y curso de la enfermedad
El diagnóstico y el curso de la enfermedad no son uniformes para las diferentes formas de ciliopatías. Los síntomas, así como el diagnóstico y el pronóstico, diferían considerablemente entre sí. Lo que tienen en común las diversas ciliopatías es que el diagnóstico se puede confirmar mediante pruebas de biología molecular.
Complicaciones
Como regla general, los afectados por la ciliopatía padecen varios síndromes diferentes y, por lo tanto, diferentes complicaciones. Estos también dependen mucho de la gravedad exacta de la enfermedad, por lo que normalmente no es posible hacer una predicción general. En primer lugar, la ciliopatía conduce a una infección en las vías respiratorias.
Esto provoca dificultad para respirar y posiblemente falta de aire. La calidad de vida de la persona afectada está significativamente restringida, por lo que ya no es posible realizar actividades extenuantes o deportivas sin más. La enfermedad también retrasa el desarrollo infantil.
En muchos casos, los afectados también sufren muy a menudo de inflamación en la nariz o las vías respiratorias. La mayoría de los afectados también carecen de la capacidad para reproducirse, por lo que también sufren trastornos psicológicos o depresión.
No existen complicaciones particulares en el tratamiento de la ciliopatía. Sin embargo, no es posible una curación completa, por lo que los afectados suelen depender siempre del uso de antibióticos y otras drogas en sus vidas.
¿Cuándo deberías ir al médico?
Si se diagnostica una enfermedad hereditaria en la familia, se debe buscar la cooperación de un médico antes de planificar un posible hijo. Los padres potenciales deben informarse exhaustivamente con anticipación sobre los riesgos o posibles desarrollos. También debe trabajar en estrecha colaboración con un médico durante el embarazo. Los exámenes preventivos ofrecidos deben realizarse para poder reaccionar de forma rápida y completa ante posibles deterioros de salud.
Dado que no se pueden usar terapias causales para enfermedades hereditarias, la intervención temprana es particularmente importante. Si no se tiene conocimiento de una mutación genética relacionada con la familia, las anomalías a menudo solo las notan los miembros del equipo obstetra inmediatamente después del nacimiento. En un proceso de rutina, se realizan los exámenes necesarios para lograr un diagnóstico. A más tardar en el desarrollo del niño, se pueden observar irregularidades en comparación con los compañeros.
Consulte a un médico en caso de cambios ópticos, trastornos del crecimiento o anomalías mentales. Las anomalías, las peculiaridades de la reacción y las irregularidades en las secuencias de movimiento deben ser examinadas por un médico. La ciliopatía es un término genérico para varios trastornos. Cada uno de ellos presenta características individuales en el paciente, por lo que se debe iniciar una visita de control con un médico si existe la sospecha de una discrepancia de salud existente.
Tratamiento y Terapia
Las terapias causales no están disponibles, las enfermedades no se pueden curar, solo se alivian con una terapia sintomática. Las terapias para las diversas ciliopatías también son diferentes.
prevención
Estas son enfermedades genéticas. Por tanto, no es posible una prevención.
Cura postoperatoria
La atención de seguimiento de la ciliopatía se basa principalmente en el tipo y la gravedad de la enfermedad. Enfermedades como el síndrome de Laurence-Moon-Biedl-Bardet o el síndrome de Joubert no se pueden curar. La atención de seguimiento se centrará en revisar los síntomas tratables y reajustar la medicación. Dado que los pacientes a menudo padecen enfermedades crónicas, se realizan exámenes de seguimiento periódicos.
Las quejas que se hayan curado, como el dolor crónico o los síntomas de intoxicación, deben tratarse con medicamentos. Si la ciliopatía se basa en una afección tratable, como un quiste renal, la atención de seguimiento depende del curso de la enfermedad y del éxito de la terapia. Si el resultado es positivo, se puede trasplantar el riñón.
Después de dicho trasplante de riñón, se lleva a cabo un examen a intervalos semanales en cooperación con el centro de trasplante y el médico de familia o especialista. Posteriormente, los intervalos se pueden reducir a cuatro veces al año. Parte de la atención de seguimiento es la determinación de valores sanguíneos, exámenes radiológicos como CT o MRI, así como otros exámenes según la enfermedad subyacente.
Al mismo tiempo, siempre se comprueba el estado general del paciente. Si la ciliopatía se debe a otras enfermedades, se debe consultar al especialista correspondiente. En cualquier caso, un especialista en enfermedades renales y el médico de familia forman parte del equipo médico.
Puedes hacerlo tu mismo
Las ciliopatías pueden adoptar una amplia variedad de formas y siempre deben tratarse individualmente. Las medidas generales que pueden promover la recuperación son la fisioterapia y un cambio de estilo de vida. Formas como el síndrome de Joubert o el síndrome de Laurence-Moon-Bardet-Biedl siempre se tratan sintomáticamente, por lo que el paciente debe comer de manera saludable, moverse lo suficiente, pero cuidarse a sí mismo. Esto al menos alivia los síntomas.
Si se ha prescrito medicación, se debe prestar especial atención a los efectos secundarios e interacciones, ya que pueden tener un efecto negativo en el desarrollo de otras ciliopatías. Si surgen complicaciones, se debe informar al médico sobre ellas. Los pacientes deben llevar un diario de quejas y anotar en detalle todos los síntomas y quejas que se noten.
El médico responsable debe decidir en cada caso qué medidas son útiles en la ciliopatía. Debido a las diversas formas de sufrimiento, siempre es necesario un plan de tratamiento individual. Se recomienda a los afectados que se pongan en contacto con su médico de cabecera, quien puede brindar más consejos sobre la mejor manera de apoyar el tratamiento médico. Además, puede establecer contacto con grupos de autoayuda que pueden proporcionar al paciente más medidas.

.jpg)


.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)