los Síndrome de West es una forma maligna generalizada de epilepsia difícil de tratar. Ocurre en bebés entre las edades de tres y doce meses.
¿Qué es el síndrome de West?
los Síndrome de West lleva el nombre del médico y cirujano inglés William James West. Observó los primeros ataques de epilepsia de este tipo en su hijo de cuatro meses en 1841 y luego describió la enfermedad desde un punto de vista científico. Como sinónimo del término Síndrome de West son también las expresiones epilepsia maligna infantil o Epilepsia BNS como abreviatura de Epilepsia de Blitz Nick Salaam usado.
Se cree que la epilepsia maligna infantil se basa en el daño cerebral orgánico que ocurrió antes, durante o después del nacimiento. Las convulsiones epilépticas generalizadas son características del síndrome de West. La enfermedad afecta a 1 de cada 4.000 a 6.000 niños. Los niños se ven afectados con más frecuencia que las niñas.
En el 90 por ciento de los niños enfermos, las convulsiones ocurren por primera vez dentro de los primeros doce meses después del nacimiento. El pico de manifestación es en el quinto mes. En casos más raros, las convulsiones no ocurren hasta la edad de dos o cuatro años. Uno de cada 20 casos de epilepsia en la primera infancia se debe al síndrome de West.
causas
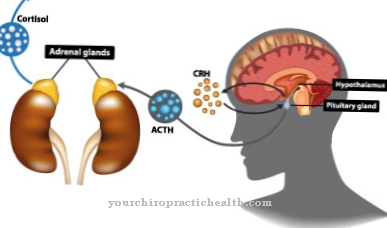
Los mecanismos exactos de desarrollo bioquímico del síndrome de West aún no se han explicado. Las convulsiones probablemente se deben a un trastorno de neurotransmisores. La causa probablemente sea un trastorno regulador del metabolismo del GABA. Una sobreproducción de la hormona liberadora de corticotropina en la glándula pituitaria también puede ser la causa. También es concebible una interacción multifactorial en el desarrollo de la enfermedad.
Dado que el síndrome de West ocurre solo en bebés y niños pequeños, el estado de madurez del cerebro parece jugar un papel en el desarrollo de las convulsiones. En los cerebros inmaduros de los recién nacidos, no todas las fibras nerviosas están todavía mielinizadas. Por lo tanto, el cerebro puede reaccionar al estrés o al daño con el síndrome de West. Se puede detectar un trastorno cerebral orgánico en dos tercios de los niños.
Se pueden encontrar trastornos del desarrollo de la corteza cerebral, microcefalia, lisencefalia o malformaciones de los vasos sanguíneos. El síndrome de Aicardi, las enfermedades cerebrales degenerativas generales, las facomatosis como la esclerosis tuberosa o la atrofia cerebral también pueden conducir al síndrome de West.
El síndrome de West también puede desarrollarse después de encefalitis o meningitis bacteriana. Otros factores de riesgo son las infecciones congénitas, las enfermedades neurometabólicas o la hipoglucemia.En la literatura especializada también se mencionan como causas el daño cerebral por hemorragia cerebral, accidente cerebrovascular, traumatismo craneoencefálico o hipoxia durante el parto.
Hay casos de enfermedades que aparecen por primera vez como efecto secundario concebible tras múltiples vacunas contra el sarampión, la rubéola y las paperas. Sin embargo, el síndrome de West aún no ha sido reconocido como un daño de la vacuna. Si se puede probar una causa, es el síndrome de West sintomático. Si no se puede detectar el síndrome de West, se supone que es un síndrome de West criptogénico. No se puede identificar ninguna causa en el 20 por ciento de todos los niños con síndrome de West.
Síntomas, dolencias y signos
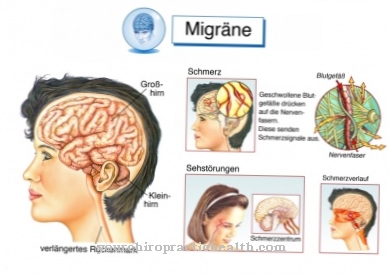
Las convulsiones epilépticas que ocurren en niños con síndrome de West se pueden dividir en tres formas diferentes. Los ataques de rayos se expresan mediante sacudidas similares a rayos de partes individuales del cuerpo o de todo el cuerpo. Las piernas se doblan de repente y los niños muestran violentas sacudidas mioclónicas.
Los músculos del cuello y la garganta se contraen durante los ataques de asentir. La barbilla se dobla hacia el pecho en un instante. La cabeza también puede estar retraída. Estos movimientos recuerdan a un movimiento de cabeza, razón por la cual las convulsiones se conocen como ataques de asentimiento.
Las convulsiones de salaam se refieren a la rápida inclinación de la cabeza y la parte superior del cuerpo hacia adelante. Al mismo tiempo, los niños levantan los brazos doblados y / o juntan las manos frente al pecho. Dado que este tipo de convulsión recuerda al saludo de Salaam, las convulsiones se denominaron ataques de Salaam.
No se puede encontrar ninguna conexión entre las convulsiones y los estímulos externos. Las convulsiones a menudo ocurren poco antes de quedarse dormido o inmediatamente después de despertarse. Clásicamente, los calambres comienzan débiles y luego aumentan más tarde en grupos de hasta 150 ataques, con menos de 60 segundos transcurridos entre los ataques individuales.
Los calambres individuales pueden variar en duración e intensidad según el niño. No hay dolor asociado con ellos y los niños generalmente permanecen completamente conscientes. Sin embargo, las convulsiones son muy agotadoras, por lo que los niños pueden llorar mucho después de una serie de convulsiones.
Diagnóstico y curso de la enfermedad
Incluso antes de que se haga el diagnóstico, los niños afectados destacan por un retraso en el desarrollo psicomotor. Se realiza un EEG para confirmar el diagnóstico. Aquí la actividad epiléptica se manifiesta en forma de ondas delta irregularmente altas y lentas. En estas ondas delta se construyen picos y ondas agudas.
Además de medir la actividad eléctrica, la sangre y la orina se examinan en el laboratorio en busca de peculiaridades cromosómicas, enfermedades hereditarias, enfermedades infecciosas y enfermedades metabólicas. Se pueden utilizar métodos de imagen como la ecografía, la tomografía por emisión de positrones, la tomografía por resonancia magnética o la tomografía computarizada para comprobar las peculiaridades del cerebro.
Complicaciones
En el peor de los casos, el síndrome de West puede provocar la muerte. Sin embargo, esto solo ocurre si la afección no se trata. Los afectados sufren ataques epilépticos a una edad muy temprana. Estos representan un peligro mortal para el niño y, por lo tanto, deben ser tratados por un médico de inmediato.
Además, la mayoría de los niños sufren espasmos, por lo que pueden producirse acoso o burlas, especialmente a una edad temprana. Esto a menudo conduce a quejas psicológicas o depresión. Asimismo, los pacientes a menudo sufren de movilidad restringida o trastornos de concentración, por lo que el desarrollo infantil también se ve significativamente restringido por el síndrome de West.
En la edad adulta, los afectados también sufren graves restricciones y trastornos. Las convulsiones epilépticas también se asocian a menudo con un dolor intenso. En muchos casos, los padres o familiares también sufren graves problemas psicológicos o depresión.
El tratamiento del síndrome de West se puede realizar con la ayuda de intervenciones quirúrgicas. No se producen compilaciones. Sin embargo, no es posible predecir si los ataques epilépticos se reducirán por completo. En muchos casos esto reduce significativamente la esperanza de vida de la persona afectada.
¿Cuándo deberías ir al médico?
La salud general de los recién nacidos y los bebés siempre debe controlarse y monitorearse regularmente. Especialmente en las primeras semanas o meses de vida, los desarrollos del niño deben ser observados y documentados lo mejor posible. Cualquier anomalía y cambio debe discutirse con el pediatra para que se pueda aclarar si es necesario actuar o si todo corresponde a un desarrollo natural.
En el caso de una convulsión o espasmos involuntarios de la descendencia, existe una necesidad urgente de actuar. Se deben iniciar exámenes médicos para aclarar la causa. Si los movimientos del niño son irregulares o no se corresponden con las condiciones naturales, es recomendable consultar a un médico. El llanto, la negativa a comer o los trastornos del tracto digestivo son señales de alerta del organismo.
Se debe consultar a un médico para poder evaluar mejor las observaciones. En caso de alteración del conocimiento o pérdida del conocimiento, se debe alertar a un servicio de emergencia. Es una situación aguda en la que se debe reaccionar al niño lo más rápido posible y se requiere atención médica intensiva. Hasta que llegue el médico de urgencias, se deben seguir las instrucciones del servicio de rescate para salvar la vida del bebé. También se debe consultar a un médico si el niño continúa llorando, cambios en la textura de la piel o sospecha que la descendencia puede tener dolor.
Terapia y tratamiento
El síndrome de West es muy difícil de tratar. Un diagnóstico temprano aumenta la probabilidad de que queden pocos o ningún daño consecuente. Si la enfermedad se basa en una peculiaridad cerebral orgánica tratable, se puede realizar una corrección quirúrgica.
Con la cirugía de la epilepsia, se pueden eliminar las causas de las convulsiones. En la mayoría de los casos, sin embargo, el síndrome de West se trata con medicamentos. Los niños reciben ACTH, corticosteroides orales o vigabatrina. También se administran Sultiam o piridoxina. Sin embargo, se ha descubierto que la mayoría de los fármacos anticonvulsivos son ineficaces en el síndrome de West.
prevención
La patogenia exacta del síndrome de West aún no está clara, por lo que la enfermedad no se puede prevenir actualmente.
Cura postoperatoria
El síndrome de West es una forma grave de epilepsia que puede tratarse con medicamentos o cirugía, por ejemplo. La administración de medicamentos como valproato o zonisamida debe estar estrictamente controlada. Los niños, en particular, son sensibles a los ingredientes activos, por lo que se recomienda encarecidamente un control estricto por parte del médico.
Los cambios repetidos de medicación son comunes para tratar la afección. La dosis debe ajustarse regularmente o la preparación debe cambiarse. Si una dieta cetogénica es parte de la terapia, el progreso debe discutirse con un especialista o nutricionista a intervalos regulares. Por lo general, se requieren más visitas al médico después de la cirugía de la epilepsia, ya que es un procedimiento riesgoso que puede tener efectos secundarios.
La frecuencia de los controles médicos depende del tipo y la gravedad de la epilepsia y del curso del procedimiento. Los padres de los niños afectados se comunican con el pediatra responsable y discuten los detalles con ellos. La atención de seguimiento la brinda el pediatra o neurólogo, que ya se está haciendo cargo de la terapia. Por lo general, la epilepsia no se puede curar de forma permanente. Por lo tanto, el cuidado posterior es un proceso continuo diseñado para curar los síntomas individuales y controlar la medicación.
Puedes hacerlo tu mismo
Los niños que padecen el síndrome de West necesitan apoyo en la vida cotidiana, ya que los ataques epilépticos recurrentes pueden ser una gran carga. Deben tomarse medidas para evitar caídas y accidentes durante un ataque epiléptico. Además, se deben agotar las opciones de tratamiento, como la cirugía de la epilepsia o el tratamiento farmacológico con vigabatrina o corticosteroides orales. Los padres de los niños afectados deben comunicarse con un centro especializado adecuado en una etapa temprana, ya que las posibilidades de recuperación disminuyen a medida que el niño crece.
Además, se aplican medidas generales para mejorar el bienestar del niño. El ejercicio físico y el apoyo mental son importantes, pero también una dieta adaptada y terapias especialmente diseñadas. Por ejemplo, se ha demostrado que una dieta cetogénica es eficaz en la epilepsia. La Asociación para la Nutrición en la Epilepsia FET e. V. da a los afectados más recomendaciones sobre la dieta.
Los niños que padecen el síndrome de West deben ser informados sobre su condición en una etapa temprana. Para ello son ideales las conversaciones con médicos y otras personas afectadas, pero también material informativo como libros o folletos. Junto con el médico responsable, se pueden desarrollar más estrategias para tratar la enfermedad a diario.
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)