los Mucopolisacaridosis es un término colectivo para las enfermedades por almacenamiento lisosómico que se basan en el almacenamiento de glicosaminoglicanos. Todas las enfermedades desarrollan síntomas y formas similares. La gravedad de los síndromes varía mucho.
¿Qué es la mucopolisacaridosis?

© Sebastian Kaulitzki - stock.adobe.com
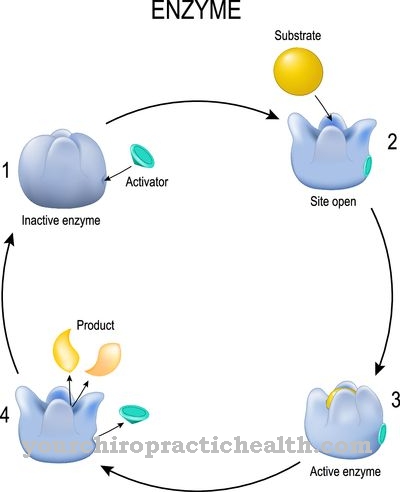
UNA Mucopolisacaridosis no existe una sola enfermedad. El término mucopolisacaridosis es un término colectivo para un gran número de enfermedades por almacenamiento, que se basan en trastornos del almacenamiento de glicosaminoglicanos (GAG) en los lisosomas de las células. El almacenamiento se realiza de forma progresiva porque la terminación de las conexiones no funciona.
Todas las mucopolisacaridosis son genéticas. Cada enfermedad carece de una enzima específica que cataliza la descomposición del GAG correspondiente. Todas las mucopolisacaridosis son enfermedades muy raras y a menudo muestran cursos similares. Si no se trata, los depósitos cada vez mayores destruyen las células. Los órganos se destruyen en el proceso. La enfermedad puede comenzar tanto en la infancia como en la edad adulta.
La mucopolisacaridosis puede ser causada por cuatro grupos diferentes de glicosaminoglicanos:
- Sulfato de heparán
- Sulfato de queratán
- Sulfato de condroitina
- Dermatan sulfato.
Todos los glicosaminoglicanos constan de una cadena de polisacárido unida a una proteína. El componente de carbohidratos constituye el 95 por ciento y el componente de proteína el cinco por ciento de la masa molecular. Dependiendo de qué glicosaminoglicano y qué enzima se vea afectada, se puede distinguir entre seis formas principales diferentes de mucopolisacaridosis: Éstas incluyen la enfermedad de Hurler / Scheie (MPS I), la enfermedad de Hunter (MPS II), la enfermedad de Sanfilippo (MPS III), la enfermedad de Morquio ( MPS IV), enfermedad de Maroteaux-Lamy (MPS VI) y enfermedad de Sly (MPS VII). Todos los tipos tienen formas leves y graves.
causas
La causa de todas las mucopolisacaridosis es un almacenamiento creciente de glicosaminoglicanos (GAG) en los lisosomas de las células. Se altera la degradación de los biopolímeros correspondientes. Para cada trastorno individual, falta una determinada enzima o esta enzima funciona incorrectamente. Puede haber varias mutaciones para cada enzima. La herencia de la mutación correspondiente puede ser autosómica recesiva, autosómica dominante o recesiva ligada a X.
Dado que un proceso enzimático generalmente implica varios pasos de reacción, teóricamente se pueden mutar varias enzimas para el mismo glicosaminoglicano. Los síntomas del trastorno serían los mismos o similares.
- A MPS I, Enfermedad de Hurler o Scheie, la enzima alfa-l-iduronidasa es defectuosa.
- MPS II representa el síndrome de Hunters con una iduronato-2-sulfatasa defectuosa.
- El síndrome de Sanfilippo (MPS III) se puede dividir en varios subtipos. Varias enzimas pueden verse afectadas en esta condición.
- Enfermedad de Morquio (MPS IV) es causada por una β-galactosidasa defectuosa.
- En el síndrome de Maroteaux-Lamy (MPS VI) es N-acetil-galactosamina-4-sulfato sulfatasa.
- La enfermedad de SlyMPS VII) está causado por una β-glucuronidasa defectuosa. Cuando los correspondientes glicosaminoglicanos se almacenan en los lisosomas, estos se vuelven cada vez más grandes.
Las células también se agrandan porque necesitan cada vez más espacio para los GAG no degradados. Esto también se nota en el agrandamiento de muchos órganos. Un síntoma típico es el agrandamiento constante del hígado y el bazo. Si no se tratan, las enfermedades por almacenamiento provocan la muerte debido a la destrucción gradual de los órganos.
Síntomas, dolencias y signos.
Los síntomas son similares para todas las enfermedades. Hay formas leves y graves. Sin embargo, un curso leve solo significa que la enfermedad progresa más lentamente. El curso final es siempre el mismo. Hay deformación progresiva del sistema esquelético, contracturas articulares, engrosamiento de los rasgos faciales y agrandamiento del hígado y el bazo.
Las habilidades mentales y motoras disminuyen a corto o largo plazo. En las formas graves de los trastornos, los cuadros clínicos son muy similares. Las hernias umbilicales e inguinales, los problemas cardíacos y las infecciones respiratorias ocurren en una etapa temprana. Con el tiempo, el estrechamiento de las vías respiratorias y el agrandamiento de las amígdalas y amígdalas se convierten en problemas masivos de apnea del sueño.
Diagnóstico y curso de la enfermedad.
Las mucopolisacaridosis se pueden diagnosticar examinando la orina en busca de glicosaminoglicanos excretados. En mucopolisacaridosis, los valores siempre aumentan. También se puede determinar la actividad de la enzima supuestamente defectuosa en los leucocitos o fibroblastos. Un cierto patrón de excreción de los glicosaminoglicanos lleva a sospechar una enzima correspondiente, que luego se examina.
Complicaciones
Debido a la mucopolisacaridosis, los afectados padecen diversas malformaciones y molestias esqueléticas. Se producen deformaciones que pueden limitar significativamente la vida diaria del paciente. Como regla general, las articulaciones también se ven afectadas por la mucopolisacaridosis, por lo que el movimiento del paciente está restringido.
Los niños en particular se ven afectados y sufren de un desarrollo muy retrasado, por lo que también pueden ocurrir varios daños consecuentes en la edad adulta. No es raro que la mucopolisacaridosis cause problemas cardíacos o respiratorios. En el peor de los casos, la muerte súbita cardíaca puede provocar la muerte de la persona afectada. Debido a las dificultades respiratorias, los pacientes sufren de cansancio y fatiga.
La capacidad de recuperación de los afectados también disminuye enormemente. No es raro que las dificultades respiratorias nocturnas provoquen problemas para dormir y, por tanto, depresión. La mucopolisacaridosis reduce significativamente la calidad de vida del paciente. Lamentablemente, no es posible un tratamiento causal de esta enfermedad. Por tanto, los afectados dependen de donantes de médula ósea para tratar los síntomas. No hay complicaciones particulares. Sin embargo, en la mayoría de los casos, los pacientes dependen de una terapia de por vida.
¿Cuándo deberías ir al médico?
Los cambios y anomalías en la estructura corporal indican un deterioro de la salud. La visita al médico es necesaria tan pronto como surjan peculiaridades ópticas permanentes o la persona en cuestión tenga problemas para optimizar deliberadamente su postura.La hinchazón de las articulaciones, los cambios en los rasgos faciales o el agrandamiento del pecho deben ser examinados intensamente por un médico para poder hacer un diagnóstico. Se requiere un médico si existen restricciones en las posibilidades de movimiento, irregularidades en el control voluntario diario y una disminución en el rendimiento físico y mental. Consulte a un médico en caso de alteraciones del ritmo cardíaco, problemas respiratorios o interrupciones durante el sueño nocturno.
La hinchazón de la garganta, la sensación de opresión en la garganta, las alteraciones en el acto de tragar y los cambios en la vocalización se consideran preocupantes. Deben ser examinados por un médico para que los síntomas puedan aliviarse. Si la persona en cuestión sufre más infecciones, si la capacidad de concentración y atención disminuye o si las hernias umbilicales o inguinales ocurren repetidamente, un médico debe ser informado de las observaciones.
Deben examinarse y tratarse las imperfecciones cutáneas repentinas, la coloración amarillenta de la piel y la inquietud interior. Se necesita un médico tan pronto como haya dolor en el cuerpo, una calidad de vida reducida y problemas de comportamiento. Si existe riesgo de dificultad para respirar, se requiere un servicio de ambulancia. Para prevenir esta afección aguda, se debe consultar a un médico lo antes posible.
Terapia y tratamiento
Hoy en día todavía no es posible una terapia causal. Sin embargo, existen algunos enfoques para la futura terapia génica para estas enfermedades en proyectos de investigación. Desafortunadamente, actualmente no hay resultados tangibles en esta área. Sin embargo, en Barcelona se iniciará un estudio clínico sobre terapia génica para la enfermedad de Hurler. Con algunas formas de mucopolisacaridosis, las transferencias de médula ósea han demostrado ser eficaces en casos individuales. Esto afecta, por ejemplo, la enfermedad de Hunter, la enfermedad de Hurler o la enfermedad de Sanfilippo.
A través de esta transferencia de médula ósea, las células madre enfermas se intercambian por células madre sanas de un donante. Esto permite al organismo restaurar suficientemente la enzima faltante. La terapia de reemplazo enzimático también vale la pena en muchos casos. Sin embargo, esta terapia de reemplazo debe llevarse a cabo de por vida. Sin embargo, también hay casos en los que las terapias prometedoras ya no son posibles. Sin embargo, el punto aquí es realizar tratamientos sintomáticos.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el dolorOutlook y pronóstico
El desarrollo posterior en pacientes con mucopolisacaridosis debe evaluarse individualmente. Este término es un término colectivo para varias enfermedades por almacenamiento. Estos están presentes en diferentes grados en cada paciente y su intensidad se pronuncia individualmente. Si no se instituye la atención médica, los órganos internos de todos los afectados se destruyen gradualmente a lo largo de su vida. Esto da como resultado un acortamiento de la vida media esperada.
Con un diagnóstico temprano, se puede elaborar una terapia optimizada personalmente. Esto está relacionado con los requisitos de salud y las quejas existentes del paciente. El tratamiento a largo plazo es fundamentalmente necesario para lograr una mejora estable de la salud. Pueden ocurrir intervenciones quirúrgicas, cada una de las cuales está asociada con diferentes riesgos y efectos secundarios. Si la operación continúa sin más complicaciones, los síntomas generalmente se alivian después.
Sin embargo, pueden ocurrir desarrollos y retrocesos no deseados en el curso de la vida. En casos individuales, solo un trasplante de médula ósea puede mejorar la calidad de vida general. Debido a las circunstancias generales, el paciente experimenta una fuerte carga emocional y mental. La vida cotidiana normal a menudo no es posible debido a los síntomas. Pueden ocurrir complicaciones psicológicas y conducir a un mayor deterioro de la situación.
prevención
Dado que las mucopolisacaridosis son enfermedades hereditarias, la prevención no es posible. En el caso de una enfermedad existente, el éxito del tratamiento puede garantizarse mediante una terapia oportuna. Además, es necesario un control constante de la función pulmonar y cardíaca. Si ya se han producido casos de mucopolisacaridosis en la familia, el riesgo de la enfermedad se puede evaluar mediante asesoramiento genético si la familia desea tener hijos.
Cura postoperatoria
En el caso de la mucopolisacaridosis, en la mayoría de los casos, el paciente solo tiene unas pocas opciones para la atención de seguimiento, por lo que la persona afectada debe principalmente consultar a un médico desde el principio. Solo con la detección temprana y el tratamiento de esta enfermedad se pueden prevenir más complicaciones, por lo que se debe contactar a un médico tan pronto como aparezcan los primeros signos y síntomas.
En la mayoría de los casos, los afectados dependen de intervenciones quirúrgicas, que pueden aliviar y limitar los síntomas. Sin embargo, dado que la mucopolisacaridosis es una enfermedad genética, generalmente no se puede curar por completo.
Por lo tanto, el interesado debe consultar primero a un médico si desea tener hijos para prevenir la recurrencia de esta enfermedad en los niños. A menudo, es muy importante contar con el apoyo de la familia durante el tratamiento. Esto también puede prevenir la depresión y otros trastornos psicológicos. La mucopolisacaridosis puede conducir a una reducción de la esperanza de vida de la persona afectada, por lo que la evolución posterior depende en gran medida del momento del diagnóstico.
Puedes hacerlo tu mismo
Las posibilidades de autoayuda en el caso de la mucopolisacaridosis se limitan a aliviar los síntomas y así mejorar la calidad de vida. Los grupos de autoayuda han demostrado ser muy útiles, ya que el intercambio con otros padres revela valiosos consejos y, a menudo, pueden aliviar temores e inquietudes y brindar una visión más positiva del futuro.
El acompañamiento a fisioterapia, terapia ocupacional, logopedia y otras formas de terapia, que a menudo se pueden profundizar en el entorno del hogar, son ahora una parte integral de la vida.
Para hacer la vida lo más fácil posible para usted y el niño afectado, es aconsejable que el entorno de vida sea accesible para los discapacitados lo antes posible. Con el aumento de la edad y el peso del niño, las camas de cuidado de altura ajustable demuestran ser un gran alivio físico para el cuidador. Los dispositivos de alerta de epilepsia y otras ayudas técnicas permiten la mayor seguridad posible incluso de noche y alivian a los padres por la noche para que puedan dormir más relajados.
Llevar un diario de síntomas puede ayudar al médico a identificar nuevos síntomas y posiblemente a corregir el tratamiento de los síntomas existentes, ya que la terapia con medicamentos a menudo no muestra el efecto deseado, sino el efecto contrario.
Dado que la enfermedad es muy exigente para los familiares, tienen que crear pequeños espacios para ellos mismos para recargar las pilas. Esto puede incluir curas, cuidados preventivos o, más tarde, unas vacaciones en el hospicio.

.jpg)
.jpg)

.jpg)




.jpg)



.jpg)