los Síndrome de Bardet-Biedl, además Síndrome de Laurence-Moon-Biedl-Bardet (LMBBS), es una enfermedad del campo de las ciliopatías que se produce exclusivamente por herencia. El síndrome se manifiesta en forma de múltiples malformaciones que se desencadenan por cambios (mutaciones) en diferentes ubicaciones de genes o cromosomas.
¿Qué es el síndrome de Bardet-Biedl?

© Creativa Images - stock.adobe.com
El cuadro clínico definido por los médicos Moon y Laurence y más tarde por Bardet y Biedl es una enfermedad en la que la distrofia retiniana se presenta como una característica médicamente significativa en combinación con otros síntomas. Debido a esta complicada situación médica inicial, la determinación final de la enfermedad de BBS es difícil. Este cuadro clínico se registró médicamente por primera vez en 1866.
Cuatro personas examinadas tenían retinitis pigmentosa (distrofia retiniana, RP) junto con paraplejía (parálisis espástica), así como hipogenitalismo (órganos genitales subdesarrollados) y una discapacidad mental. En 1920 el médico francés Bardet describió una enfermedad que se componía de RP (distrofia retiniana), hipogenitalismo, polidactilia y obesidad.
El patólogo de Praga Biedl también encontró debilidad (confusión mental). En 1925, los investigadores Weiss y Solis-Cohen resumieron los casos conocidos y describieron el cuadro clínico como Síndrome de Laurence-Moon-Biedl-Bardet.
causas
En los años siguientes, la literatura médica señaló cada vez más que los casos registrados por Laurence y Moon son una forma especial rara que solo ocurre en casos aislados junto con BBS. Los resultados de investigaciones médicas más recientes asignan el síndrome de Bardet-Biedl al área de las ciliopatías (enfermedades ciliares).
Estas enfermedades muestran un mal funcionamiento común de los llamados cilios (pequeños apéndices, antenas) que aparecen en la mayoría de las células del organismo humano. Las ciliopatías se caracterizan por transiciones fluidas y superposiciones entre diferentes enfermedades ciliares.
Síntomas, dolencias y signos
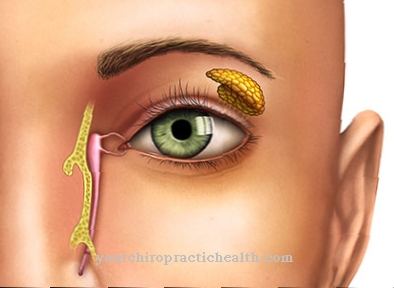
La característica principal de la distrofia retiniana hereditaria es un término genérico que describe el inicio de la pérdida de función y la posterior degeneración (destrucción) de los fotorreceptores. Conducen a una pérdida progresiva (progresiva) de la función visual. Las alteraciones visuales que progresan rápidamente suelen aparecer muy temprano en los niños, cuando tienen entre cuatro y diez años. Se hacen sentir de diferentes formas, dependiendo de los fotorreceptores afectados.
Como una "forma de cono de varilla" con el curso característico de la retinosis pigmentaria (RP), la enfermedad tiene su origen en la periferia de la retina (retina externa) y se convierte en degeneración macular (destrucción de la vista aguda) a través de una pérdida progresiva del campo de visión.
Con la obesidad (obesidad), el cuerpo muestra una acumulación patológica de tejido graso. En el caso del BBS, el aumento anormal de la acumulación de grasa en las piernas, el estómago, las nalgas, los brazos, el pecho y las caderas se produce predominantemente como obesidad del tronco, y el tronco, las piernas y los muslos se ven especialmente afectados. La polidactilia es un síntoma notable y una característica importante del síndrome de Bardet-Biedl. El hallazgo no es fácil porque la polidactilia rudimentaria se corrige quirúrgicamente después del nacimiento.
Los rayos X pueden proporcionar más información. La polidactilia puede aparecer con diferentes signos, por ejemplo, como un dedo rudimentario o un apéndice de un dedo. Se puede formar un dedo del pie o de la mano adicionalmente o solo parcialmente. La hexadactilia unilateral en el pie y / o la mano tiene un vínculo adicional, la hexadactilia bilateral ocurre en ambos pies y / o manos.
Los dedos de los pies o de las manos que han crecido juntos (sindactilia) y el acortamiento de uno o más dedos o dedos de las manos (braquidactilia) también son signos de BBS. Solo unos pocos pacientes tienen las cuatro extremidades afectadas. El retraso del desarrollo mental es diferente. Solo un pequeño número de los afectados presenta retraso mental severo. Es posible una inteligencia normalmente entrenada.
Los niños aprenden a hablar y caminar tarde y, en ocasiones, presentan problemas de conducta como trastornos de ansiedad. Los comportamientos compulsivos o autistas, un bajo umbral de frustración y una emocionalidad inestable son otros posibles efectos secundarios. Se prefiere lo familiar, pero se rechazan los cambios. Son comunes las anomalías en los órganos genitales internos y externos.
Otros cambios son el hipospadias (la abertura uretral está por encima o por debajo, en lugar de en la parte delantera del pene), el abdomen o los testículos inguinales, las constricciones uretrales, la constricción del prepucio y las válvulas uretrales posteriores. En las pacientes femeninas, se conoce la atresia vaginal (la vagina no está abierta), la falta de aberturas uretrales y los labios menores internos reducidos.
No es raro que las mujeres afectadas tengan ciclos menstruales irregulares. Los cambios renales son efectos secundarios comunes. El hallazgo depende del examen del tracto urinario inferior y los riñones mediante ecografía (ecografía).
Diagnóstico y curso de la enfermedad
El síndrome de Bardet-Biedl (BBS) tiene seis síntomas principales, pero no ocurren juntos en todos los casos. Los médicos asumen un hallazgo correspondiente si están presentes al menos cuatro de los síntomas principales. Alternativamente, existe una alta probabilidad de que la enfermedad esté presente si el paciente tiene tres síntomas principales y dos síntomas secundarios.
Los seis síntomas principales son distrofia retiniana, obesidad (acumulación anormal de tejido adiposo, sobrepeso), polidactilia (exceso de dedos y / o dedos), retraso mental (retraso del desarrollo mental), hipogenitalismo (órganos genitales subdesarrollados) y enfermedad renal. Los síntomas secundarios de baja frecuencia incluyen retrasos en el habla, déficit del habla, malformaciones cardíacas, ataxia (alteración de la coordinación del movimiento), asma, diabetes mellitus (diabetes), enfermedad de Crohn (inflamación del intestino grueso y / o delgado), displasia de las costillas y vértebras y cifoescoliosis (anemia vertebral) en.
Complicaciones
Con el síndrome de Laurence-Moon-Biedl-Bardet, los afectados suelen sufrir una pérdida de la función visual. La pérdida no se produce de repente, sino de forma gradual. En el peor de los casos, los afectados quedarán completamente ciegos y, por lo general, ya no podrán recibir tratamiento.
Especialmente en los jóvenes y los niños, la ceguera puede provocar graves problemas psicológicos o incluso depresión. Los pacientes están claramente restringidos en su vida diaria y sufren de un campo de visión muy reducido. En muchos casos, el síndrome de Laurence-Moon-Biedl-Bardet también conduce a problemas de comportamiento, por lo que los niños en particular pueden sufrir acoso o burlas.
El desarrollo de los niños también se ve retrasado y restringido significativamente por el síndrome. También pueden ocurrir trastornos de ansiedad. No es raro que el síndrome de Laurence-Moon-Biedl-Bardet dé lugar a quejas psicológicas y depresión en familiares o padres. Lamentablemente, no es posible un tratamiento causal del síndrome de Laurence-Moon-Biedl-Bardet.
Algunas quejas pueden ser limitadas. Sin embargo, no se establece un curso completamente positivo de la enfermedad. El síndrome no reduce la esperanza de vida del paciente. En algunos casos, los afectados a veces necesitan la ayuda de otras personas en su vida diaria.
¿Cuándo deberías ir al médico?
Dado que el síndrome de Laurence-Moon-Biedl-Bardet es una enfermedad hereditaria, el diagnóstico se puede realizar en el útero. A más tardar después del parto, se debe consultar a un médico si se notan síntomas típicos como alteraciones visuales u obesidad. Las malformaciones de los dedos de los pies y las manos también son un claro indicador de una enfermedad.Los padres que noten síntomas en su hijo deben informar al pediatra de inmediato.
Un examen completo proporciona información sobre la enfermedad. A partir de entonces, la terapia suele iniciarse directamente, que consta de diversos tratamientos por parte de ortopedistas, neurólogos, oftalmólogos, internistas y terapeutas, así como fisioterapeutas. Son necesarias más visitas al médico si el tratamiento no tiene el efecto deseado. También se requiere asesoramiento médico en situaciones de emergencia, por ejemplo, si el niño se cae como resultado de una malformación o tiene una convulsión repentina. Si la persona enferma muestra signos de malestar emocional, los padres deben consultar a un terapeuta adecuado. Los niños mayores pueden ponerse en contacto con el psicólogo de la escuela junto con sus padres y discutir las medidas adecuadas.
Terapia y tratamiento
Esta enfermedad se produce sobre la base de una herencia autosómica recesiva, lo que significa que ambas copias (alelos) de un gen BBS muestran un cambio (mutación). Los padres del paciente son de “sangre mixta” y cada uno porta un alelo modificado y otro inalterado del gen correspondiente. No tienen la enfermedad. Los niños solo se enferman si su padre y su madre transmiten el alelo mutado. Con niños adicionales, la probabilidad de repetición es del 25 por ciento.
Aún no se conoce una opción de terapia causal, ya que ciertos síntomas de la enfermedad aún no se pueden asignar de manera concluyente a los diversos cambios genéticos. Los síntomas y sus manifestaciones aparecen de manera diferente incluso en hermanos enfermos. Dado que el cuadro completo característico de BBS solo está presente en casos raros, especialmente en niños pequeños, es difícil un diagnóstico correspondiente.
Debido a los síntomas oligosintomáticos que se presentan con frecuencia, con los que ocurren muy pocos síntomas atípicos y solo levemente pronunciados, otros posibles cuadros clínicos deben tenerse en cuenta en el diagnóstico diferencial. Los cambios en el mismo gen pueden dar lugar a diferentes cuadros clínicos, por ejemplo, síndrome de Joubert, Bardet-Biedl o Meckel-Gruber.
Outlook y pronóstico
El pronóstico de la presencia del síndrome de Laurence-Moon-Biedl-Bardet es generalmente malo porque las malformaciones múltiples son congénitas e incurables. Si ocurren cuatro de los seis síntomas principales, se confirma el diagnóstico de síndrome de Laurence-Moon-Biedl-Bardet. Numerosos síntomas secundarios se suman a los síntomas principales. Esto incluye una ceguera progresiva.
Debido a la complejidad de los síntomas, no hay perspectivas de cura. Sólo existe una posibilidad mediocre de un notable alivio de los síntomas. El número de posibles malformaciones y trastornos en el síndrome de Laurence-Moon-Biedl-Bardet es tan grande que la enfermedad hereditaria es difícil de tratar. En cualquier caso, el curso de esta enfermedad genética no se puede influir. Sin embargo, los síntomas actuales se pueden aliviar parcialmente.
Sin embargo, el mal pronóstico general no reduce la esperanza de vida de las personas afectadas. A una edad avanzada y después de quedar ciego, los afectados pueden depender permanentemente de ayuda o cuidados. A través de esfuerzos médicos interdisciplinarios, muchos pacientes con síndrome de Laurence-Moon-Biedl-Bardet pueden experimentar un curso algo más leve de la enfermedad.
Los crecientes problemas visuales representan una parte problemática y difícil de tratar de la enfermedad, ya que los niños pequeños afectados padecen de problemas visuales cada vez mayores. Empeoran con el tiempo. Los problemas de visión no tienen por qué provocar ceguera en todos los afectados. Las secuelas psicológicas del síndrome de Laurence-Moon-Biedl-Bardet generalmente se pueden tratar bien.
prevención
La prevención en el sentido de prevenir esta enfermedad no es posible. Es importante realizar un seguimiento regular de los síntomas y los síntomas acompañantes. Los controles repetidos de la presión arterial y la función renal, el asesoramiento nutricional, la fisioterapia y la terapia ocupacional, así como la terapia del habla, son posibles enfoques terapéuticos.
Cura postoperatoria
En la mayoría de los casos, los afectados por el síndrome de Laurence-Moon-Biedl-Bardet no tienen opciones especiales de seguimiento disponibles, por lo que se debe contactar y consultar a un médico muy temprano en esta enfermedad. Como regla general, la autocuración no puede ocurrir, por lo que siempre es necesario el tratamiento por parte de un médico.
Dado que el síndrome de Laurence-Moon-Biedl-Bardet es una enfermedad hereditaria, la persona interesada debe someterse a un examen genético y asesoramiento si desea tener hijos para que el síndrome de Laurence-Moon-Biedl-Bardet no se transmita a su descendencia. se transmite. En muchos casos, los afectados dependen de intervenciones quirúrgicas para paliar las malformaciones y deformidades.
Aquí, la persona afectada definitivamente debe descansar después del procedimiento y cuidar su cuerpo. En cualquier caso, se debe evitar el esfuerzo u otras actividades físicas y estresantes para no sobrecargar innecesariamente el cuerpo. Dado que el síndrome de Laurence-Moon-Biedl-Bardet también puede provocar un comportamiento anormal, los padres deben apoyar y alentar al niño en su desarrollo. También son necesarias discusiones amorosas e intensas con el niño para prevenir trastornos psicológicos o depresión.
Puedes hacerlo tu mismo
El síndrome de Laurence-Moon-Biedl-Bardet tiene varios síntomas, y el paciente a menudo sufre la mayor parte de la función visual deteriorada. Incluso con los niños, la capacidad habitual de ver comienza a deteriorarse, por lo que son los padres quienes presentan al niño al médico y así agilizan el diagnóstico. De esta manera, la enfermedad se puede tratar rápidamente, aunque las opciones de tratamiento hasta ahora solo han sido sintomáticas.
Las alteraciones visuales aumentan cada vez más en los niños enfermos y por tanto perjudican considerablemente la vida cotidiana, por lo que la calidad de vida de los afectados disminuye. Porque los problemas de visión desarrollan numerosas dificultades para el paciente cuando asiste a la escuela, en su tiempo libre y en lo que respecta a su integridad física. El riesgo de accidentes también aumenta significativamente, por ejemplo, en el tráfico rodado. Es por eso que los padres acompañan a sus hijos enfermos siempre que pueden o contratan personal de enfermería para que el paciente no se quede solo.
En algunos casos, la enfermedad se propaga hasta la ceguera. Dado que tal desarrollo ya es evidente de antemano, los pacientes se preparan para ello. Los padres rediseñan el espacio vital para que no contenga ninguna fuente de peligro para la persona con discapacidad visual. Además, las víctimas ciegas aprenden a utilizar un bastón largo para poder moverse de forma independiente fuera de su propia casa.

.jpg)

.jpg)