los Síndrome de Allan-Herndon-Dudley es una mutación en el gen SLC16A2 que cambia el transportador de hormonas tiroideas MCT8 y causa una absorción deficiente de yodotironina en el tejido muscular y el sistema nervioso central. Debido a la mutación, los afectados sufren debilidad muscular y retrasos en el desarrollo móvil y mental. El AHDS es incurable y hasta ahora solo se ha tratado con la administración de triyodotiroacetato.
¿Qué es el síndrome de Allan-Herndon-Dudley?

Los retrasos en el desarrollo físico, mental o emocional de los adolescentes y los niños se resumen como retrasos o retraso en el desarrollo. Los retrasos en el desarrollo pueden tener diferentes causas. Por ejemplo, el desencadenante del retraso en el desarrollo puede estar en el sistema nervioso central.
Este es el caso, por ejemplo, del síndrome de Allan-Herndon-Dudley (AHDS). Además del retraso mental severo, el síndrome se caracteriza por trastornos del desarrollo motor. La primera descripción del síndrome se remonta a 1944. El cuadro clínico que describe es una enfermedad hereditaria, es decir, un trastorno genético.
La enfermedad afecta a los bebés varones en la mayoría de los casos documentados hasta la fecha. Los trastornos del desarrollo y sus consecuencias son en casi todos los casos claramente visibles desde el nacimiento. El AHDS es una enfermedad extremadamente rara.Por esta razón, el estado de la investigación sobre el síndrome de Allan-Herndon-Dudley hasta ahora ha sido bastante pobre.
causas
El AHDS es un trastorno genético hereditario causado por una mutación en el gen SLC16A2. Este es el gen codificador del denominado transportador de hormonas tiroideas MCT8. Este transportador media la captación de yodotironinas en el tejido muscular y nervioso.
Debido a la mutación, se producen alteraciones en la captación de hormonas tiroideas, que alteran el sistema nervioso central y, por lo tanto, perjudican el desarrollo de las células del sistema nervioso. El tejido muscular y el cerebro se empobrecen debido a la desregulación relacionada con la mutación de la hormona tiroidea activa de la que realmente dependen.
El síndrome se transmite como una herencia recesiva ligada al cromosoma X. Las mujeres pueden heredar la enfermedad, pero debido a su estructura cromosómica doble X, rara vez se enferman. Los hombres enfermos no pueden reproducirse.
Aunque la mutación es genética, además de este factor interno, los factores externos probablemente juegan un papel en el inicio de la enfermedad. Debido a la rareza y la base de investigación limitada, el papel de estos factores externos aún no se ha aclarado de manera concluyente.
Puedes encontrar tu medicación aquí
➔ Medicamentos para la debilidad muscularSíntomas, dolencias y signos
El síndrome de Allan-Herndon-Dudley es una enfermedad congénita que generalmente se manifiesta en niños pequeños o bebés. Los afectados sufren debilidad muscular más o menos severa. El tejido muscular de los niños está notablemente subdesarrollado. La debilidad de los músculos pronto se acompaña de deformidades articulares.
Las contracturas también son síntomas acompañantes frecuentes. La movilidad de los niños se ve cada vez más afectada por las contracturas y deformidades. Por esta razón, los afectados a menudo parecen anormalmente estáticos o incluso inmóviles. Debido a la falta de suministro de hormonas tiroideas relacionada con la mutación, los afectados a menudo también sufren calambres musculares o realizan movimientos involuntarios con los brazos y las piernas.
A menudo, los afectados no pueden moverse de forma independiente. En la mayoría de los casos, las deficiencias motoras están asociadas con trastornos mentales graves. Por ejemplo, la mayoría de los pacientes no pueden hablar. En casos individuales, el AHDS se puede caracterizar por muchos otros síntomas en el área del desarrollo mental y físico.
Diagnóstico y curso
El médico suele sospechar primero AHDS en la anamnesis. En las pruebas de laboratorio, un nivel elevado de T3 con niveles normales de FT4 y TSH indica síndrome de Allan-Herndon-Dudley. La obtención de imágenes del sistema nervioso central suele formar parte del diagnóstico.
Las debilidades musculares debidas a enfermedades de las neuronas motoras pueden excluirse del diagnóstico diferencial. El pronóstico para los pacientes con síndrome de Allan-Herndon-Dudley es relativamente malo. Hasta ahora, la enfermedad es incurable. Los estudios han sugerido que el momento del diagnóstico puede desempeñar un papel fundamental en el pronóstico del paciente.
Complicaciones
Como todos los trastornos hereditarios cromosómicos, el síndrome de Allan-Herndon-Dudley no se puede tratar de forma curativa. El efecto secundario más común del síndrome de Allan-Herndon-Dudley (debilidad muscular pronunciada) se puede tratar con fisioterapia. Dicho tratamiento destinado a fortalecer los músculos puede ser doloroso para el paciente.
Los niños pequeños, en particular, a menudo rechazan la terapia debido al dolor. A pesar del entrenamiento intensivo, la fisioterapia no siempre conduce al éxito deseado. Es similar con el apoyo de la terapia del habla del paciente de Allan-Herndon-Dudley. Aunque la reducción de la capacidad del lenguaje se puede mejorar con un entrenamiento intensivo, el tratamiento no siempre conduce al éxito debido al deterioro mental mayoritariamente de alto grado de los afectados.
La frustración por parte del paciente y una gran carga para toda la familia es una de las complicaciones más graves en el tratamiento del síndrome de Allan-Herndon-Dudley. Los calambres musculares y los movimientos de las extremidades que no se pueden modificar pueden tratarse con la administración de relajantes musculares. Se pueden observar complicaciones en los efectos secundarios, a veces graves, de los medicamentos.
Cabe mencionar la fatiga, una sensación general de cansancio y malestar, además del estrés en el tracto gastrointestinal. El uso prolongado de relanxantes también daña el hígado y los riñones. Si no se trata el síndrome de Allan-Herndon-Dudley, los afectados no podrán hacer ningún progreso significativo en términos de sus habilidades mentales o motoras.
¿Cuándo deberías ir al médico?
En muchos casos, no es posible el tratamiento directo del síndrome de Allan-Herndon-Dudley. Por esta razón, el tratamiento es principalmente sintomático y está dirigido a las quejas y retrasos individuales. Como regla general, los padres deben consultar a un médico si el niño tiene debilidad muscular.
Esto puede notarse por agotamiento o cansancio persistente. También es necesario el consejo médico si el síndrome de Allan-Herndon-Dudley ralentiza el desarrollo mental y motor.
Si el tratamiento no se lleva a cabo en la niñez, puede provocar molestias y restricciones considerables en la edad adulta. Se debe consultar a un médico, especialmente si el paciente ya no puede hablar. El tratamiento también es necesario para los calambres musculares. Si se trata de una emergencia aguda, puede ir directamente al hospital o llamar a una ambulancia.
En la mayoría de los casos, el síndrome de Allan-Herndon-Dudley será tratado por un médico de cabecera o un pediatra. Sin embargo, las quejas individuales deben ser examinadas y tratadas por el especialista o terapeuta correspondiente.
Doctores y terapeutas en su área
Tratamiento y Terapia
El AHDS es una enfermedad causalmente intratable. Dado que no existen terapias disponibles para corregir la causa principal, la enfermedad hasta ahora no ha sido curable. Mientras tanto, los avances en el campo de la terapia génica sugieren que los enfoques de la terapia génica pronto se aprobarán para la práctica clínica diaria.
Aún no se ha aclarado en qué medida los pacientes con el síndrome se beneficiarían de la aprobación. En la actualidad tampoco existe una opción de tratamiento establecida o estandarizada para los pacientes con AHDS en el campo de la terapia sintomática. Hace unos años los investigadores consideraron la administración de TRIAC como una posible opción de terapia sintomática.
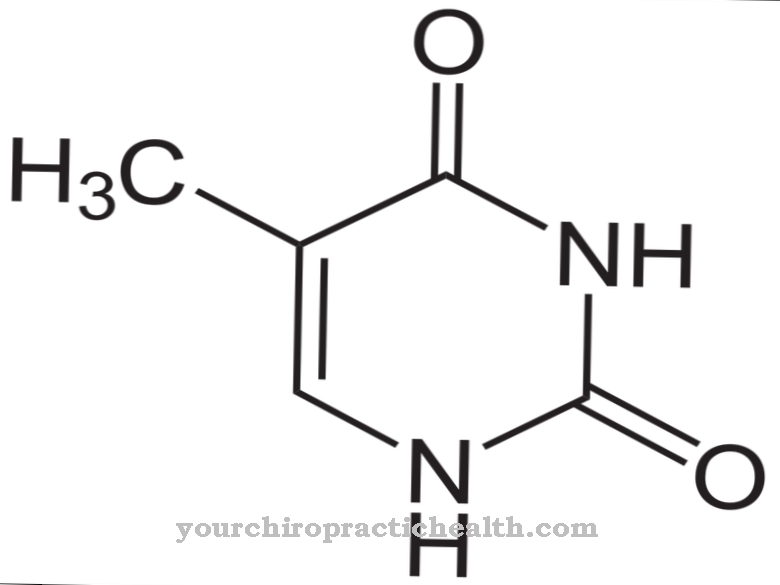
TRIAC es una hormona tiroidea no clásica, triyodotiroacetato. La administración de la hormona se llevó a cabo en un estudio clínico en niños afectados, pero no pudo lograr ningún resultado visible. Los resultados del estudio no son necesariamente significativos porque la administración de la hormona comenzó relativamente tarde.
Por esta razón, en 2014 TRIAC todavía se consideraba la mejor terapia posible. En un caso en 2014, se documentó una mejora significativa en el desarrollo motor y mental durante el tratamiento con TRIAC. La terapia se inició en la persona afectada en la primera infancia.
Por tanto, los resultados de los estudios hasta la fecha indican que el momento en el que se inicia la terapia tiene un efecto sobre los resultados de la terapia que no debe subestimarse para los pacientes con AHDS. Teóricamente, las terapias de apoyo complementarias, como la terapia ocupacional y la fisioterapia o la intervención temprana, pueden utilizarse para mejorar la calidad de vida y las habilidades de los pacientes. Sin embargo, apenas existe evidencia de la eficacia de tal enfoque en relación con los pacientes con AHDS.
Outlook y pronóstico
El síndrome de Allan-Herndon-Dudley causa varios síntomas diferentes en la mayoría de los pacientes. En primer lugar, los afectados sufren de debilidad muscular severa. Esto significa que las actividades normales o los deportes ya no se pueden realizar fácilmente para la persona en cuestión. También hay importantes retrasos en el desarrollo intelectual y móvil. La concentración del paciente está claramente restringida y reducida.
Todavía hay fuertes calambres en los músculos y, por lo tanto, a menudo movimientos involuntarios o espasmos. A medida que avanza el síndrome de Allan-Herndon-Dudley, los afectados ya no pueden hablar. La vida cotidiana del paciente se ve significativamente restringida por el síndrome y la calidad de vida se reduce. En algunos casos, los pacientes dependen de la ayuda de otras personas en su vida diaria.
Por lo general, no es posible tratar el síndrome de Allan-Herndon-Dudley de manera causal. Por esta razón, el tratamiento es solo sintomático. Los afectados dependen de diversas terapias que, sin embargo, no siempre conducen a un curso positivo de la enfermedad. En algunos casos, el síndrome de Allan-Herndon-Dudley limita la esperanza de vida de los afectados.
Puedes encontrar tu medicación aquí
➔ Medicamentos para la debilidad muscularprevención
El AHDS solo se puede prevenir mediante asesoramiento genético. Los portadores de mutaciones pueden, por ejemplo, decidir no tener hijos propios.
Cura postoperatoria
La necesidad de atención de seguimiento para el síndrome de Allan-Herndon-Dudley causado genéticamente solo afecta a los bebés varones. El problema es que no existe una forma de terapia adecuada para esta condición hereditaria. Las graves consecuencias de los defectos en los transmisores de hormonas tiroideas difícilmente pueden mejorarse. Los intentos de aliviar a los niños afectados dándoles hormonas tiroideas especiales han fracasado.
El problema es que la base de la enfermedad suele estar ya establecida en el cuerpo de la madre. Dañan permanentemente al feto. Desde este punto de vista, el tratamiento comienza demasiado tarde, es decir, solo después del nacimiento. En el cuidado posterior, solo se puede tratar el daño que ya está presente. Sin embargo, hay esperanza. En 2014, se conoció un caso en el que un bebé con síndrome de Allan-Herndon-Dudley fue tratado con éxito con TRIAC. La atención de seguimiento seguía siendo necesaria porque el niño no se podía curar. Al menos sus síntomas se aliviaron.
El síndrome de Allan-Herndon-Dudley es causado en parte por una barrera hematoencefálica defectuosa. Esto es lo que sugieren los estudios del Hospital Cedars Sinai. Las hormonas tiroideas no pueden funcionar debido a la barrera hematoencefálica defectuosa. Esto también se aplica a las hormonas tiroideas que se administran después del nacimiento del niño. La biotecnología o la investigación genética pueden ayudar. Todos los intentos de tratamiento están fracasando actualmente. Esto también afecta la atención de seguimiento para los niños gravemente dañados.
Puedes hacerlo tu mismo
El síndrome de Allan-Herndon-Dudley es una afección grave que aún no se ha tratado de manera eficaz. Sin embargo, los padres aún pueden tomar algunas medidas para apoyar la terapia.
En primer lugar, el entrenamiento cognitivo y la actividad física regulares son importantes. Una terapia integral, que, dependiendo de la gravedad del síndrome, puede consistir en entrenamiento del habla y lectura, pero también de ejercicios cerebrales generales, también ofrece ejercicios de fisioterapia. El entrenamiento debe adaptarse individualmente a los síntomas. Por lo tanto, los padres de los niños afectados deben asegurarse de que las medidas se elijan de manera óptima y que el niño no se sienta abrumado.
En el caso de trastornos mentales graves, el niño puede necesitar apoyo permanente en la vida diaria. Un servicio de atención ambulatoria puede ser un alivio importante para los padres. El tratamiento hospitalario es igualmente importante, que puede apoyarse en el hogar mediante un control regular de los síntomas.
Los padres también deben aprovechar la asesoría psicológica y, si es necesario, acudir también a un grupo de autoayuda, porque el contacto con otras personas afectadas facilita el afrontamiento de la enfermedad. Además, los padres a menudo reciben consejos importantes sobre cómo tratar a un niño enfermo.
.jpg)
.jpg)

.jpg)

.jpg)





.jpg)