los Yo enfermedad celular es una mucolipidosis liosomal. La causa de la enfermedad de almacenamiento es una mutación del gen GNPTA con el locus del gen q23.3 en el cromosoma 12. El tratamiento sintomático se lleva a cabo principalmente mediante la administración de bisfosfonatos.
¿Qué es la enfermedad de las células I?

© red150770 - stock.adobe.com
Las enfermedades por almacenamiento se caracterizan por el depósito de diversas sustancias en células y órganos del cuerpo humano. Es un grupo heterogéneo de enfermedades que se pueden dividir en varias subformas. Además de las glucogenosis, mucopolisacaridosis y lipidosis, la medicina diferencia, según la sustancia depositada, esfingolipidosis, hemosiderosis y amiloidosis.
Las enfermedades por almacenamiento lisosómico afectan a los lisosomas. Estos son pequeños orgánulos celulares recubiertos de membrana en los eucariotas. Los lisosomas están formados por el aparato de Golgi y están equipados con enzimas hidrolíticas y fosfatasas. Con la ayuda de sus enzimas, deben digerir principalmente sustancias extrañas y las propias sustancias del cuerpo.
La enfermedad de células I es una mucolipidosis liosomal con dos subtipos diferentes. Leroy y DeMars documentaron por primera vez la enfermedad en la década de 1960, señalando su similitud con la mucopolisacaridosis tipo I, conocida como enfermedad de Hurler. El nombre de la enfermedad proviene de las inclusiones de fibroblastos, las llamadas células de inclusión, en la piel del paciente.
causas
La causa de la enfermedad de las células I radica en la falta de actividad de la N-acetilglucosaminil-1-fosfotransferasa. La actividad restringida de esta enzima evita que una gran parte de las enzimas lisosomales ingresen al interior del lisosoma. La regulación de las enzimas lisosomales está determinada por la actividad de la fosfotransferasa.
Permite la síntesis de una señal de clasificación en un organismo sano. Este proceso se altera en la enfermedad de células I. Por lo tanto, no hay etiquetado con manosa-6-fosfato. Por esta razón, las enzimas lisosomales ya no se clasifican correctamente y migran a la matriz extracelular de manera incontrolada a través de la membrana plasmática.
La razón de esto es una mutación en el gen GNPTAB. Elimina la funcionalidad de la N-acetilglucosaminil-1-fosfotransferasa y, por lo tanto, la capacidad de catalizar la síntesis de manosa-6-fosfato. El transporte de enzimas lisomales está muy alterado. La N-acetil-glucosamina-1-fosfotransferasa consta de las subunidades alfa, beta y gamma. Están codificados en dos genes.
La enfermedad hereditaria de células I afecta al gen GNPTA en el cromosoma 12. Hay una mutación en el locus del gen q23.3. Para la enfermedad rara, se da una incidencia de alrededor de 0,3: 100.000. La herencia está sujeta a herencia autosómica recesiva. Por tanto, ambos padres deben ser portadores del gen defectuoso para transmitir la enfermedad.
Síntomas, dolencias y signos
En la mayoría de los casos, los síntomas de la enfermedad de células I se pueden observar inmediatamente después del nacimiento o unos meses después como muy tarde, y sus características son similares a las del síndrome de Hurler. A diferencia de los pacientes con síndrome de Hurler, aquellos con enfermedad de células I no muestran excreción de mucopolisacáridos.
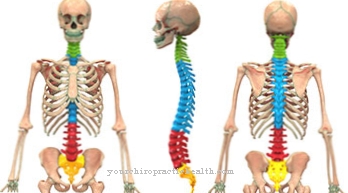
Los síntomas individuales de la enfermedad están sujetos a una gran cantidad de variaciones. Kornfeld y Sly resumen las características clínicas del esqueleto, los órganos internos, los ojos, la piel, el sistema nervioso central y la cara. El esqueleto se ve afectado con mucha frecuencia por cifoescoliosis y luxaciones de cadera.
También pueden presentarse pie zambo, contracturas articulares y deformidades de las vértebras. Lo mismo se aplica a la baja estatura y la disostosis múltiple. La enfermedad puede manifestarse en los órganos internos en forma de hepatoesplenomegalia y cardiomegalia o enfermedad cardíaca. El rostro del paciente tiene rasgos toscos.
El exoftalmos, encías hiperplásicas o escafocefalia son síntomas típicos. También son características una boca abierta y una nariz profundamente hundida. Los ojos de los afectados suelen presentar opacidades corneales o párpados hinchados. La piel es gruesa y áspera, con un severo retraso psicomotor o mental en el sistema nervioso central.
Diagnóstico y curso de la enfermedad
El primer diagnóstico de sospecha de enfermedad de células I puede realizarse mediante un diagnóstico visual basado en la anamnesis. Puede utilizarse una determinación bioquímica de la actividad enzimática lisosomal en el suero para confirmar el diagnóstico. Esta determinación revela una relación absurda entre la actividad intra y extracelular.
También se puede determinar la actividad de la fosfotransferasa en los fibroblastos para confirmar el diagnóstico. Las inclusiones corresponden a mucopolisacáridos, lípidos u oligosacáridos. Los diagnósticos genéticos moleculares pueden disipar cualquier duda restante. Si hay un historial apropiado, la enfermedad también se puede diagnosticar como parte del diagnóstico prenatal.
Debido a la baja prevalencia, la evaluación prenatal solo se recomienda en realidad si existe una disposición familiar. El curso de la enfermedad depende de los síntomas del caso individual y no es directamente predecible. La mayoría de los pacientes, sin embargo, apenas sobreviven a los diez años. Sin embargo, las formas más leves de desarrollo no están completamente excluidas en casos individuales.
Complicaciones
La enfermedad de células I puede provocar diversas complicaciones y quejas. Sin embargo, estos se reconocen tarde, por lo que la enfermedad de células I solo puede diagnosticarse tarde. Los síntomas son relativamente inconsistentes, lo que a menudo dificulta el tratamiento. Esto suele provocar molestias y malformaciones de la piel, los ojos y los órganos internos.
En el peor de los casos, la persona afectada puede quedarse ciega o morir directamente por insuficiencia orgánica. Además, hay una baja estatura pronunciada y también problemas cardíacos. Los párpados suelen estar hinchados y hay disminución de la inteligencia y retraso mental. No es raro que la persona afectada dependa de la ayuda de otras personas en la vida cotidiana debido al retraso para afrontarlo.
La calidad de vida del paciente se ve muy reducida por la enfermedad de células I. Como regla general, no existen complicaciones particulares en el tratamiento de la enfermedad. Se utilizan medicamentos y tratamientos psicológicos que pueden aliviar los síntomas. Sin embargo, no es posible un tratamiento completo y causal de esta enfermedad. La enfermedad reduce la esperanza de vida.
¿Cuándo deberías ir al médico?
La enfermedad de células I generalmente se diagnostica inmediatamente después del nacimiento del niño. La necesidad de otras medidas de tratamiento depende del tipo y la gravedad de los síntomas. Las malformaciones menores no necesariamente tienen que tratarse. El pie zambo y las deformidades de las vértebras, por otro lado, son malformaciones graves que deben tratarse quirúrgicamente y con medicación. Los padres deben consultar a un especialista inmediatamente si el médico responsable en la maternidad aún no lo ha hecho.
Si ocurre un accidente o una caída como resultado de las quejas, el niño debe ser trasladado al hospital o los padres deben llamar a los servicios de emergencia de inmediato. En el caso de malformaciones graves, que también pueden afectar la psique del niño más adelante en la vida, se debe consultar a un terapeuta para acompañar el tratamiento médico. Por lo tanto, la enfermedad de células I siempre requiere un examen médico. La persona de contacto adecuada es el pediatra o un especialista en enfermedades hereditarias. En caso de alteraciones visuales, se debe consultar a un oftalmólogo.
Doctores y terapeutas en su área
Tratamiento y Terapia
La enfermedad de las células I se considera incurable. Por tanto, no existe una terapia causal. El tratamiento es solo sintomático y de apoyo. La atención psicoterapéutica para las familias afectadas constituye una gran parte de la terapia de apoyo. La terapia sintomática depende del caso individual. Los síntomas óseos a menudo se tratan con bifosfonatos.
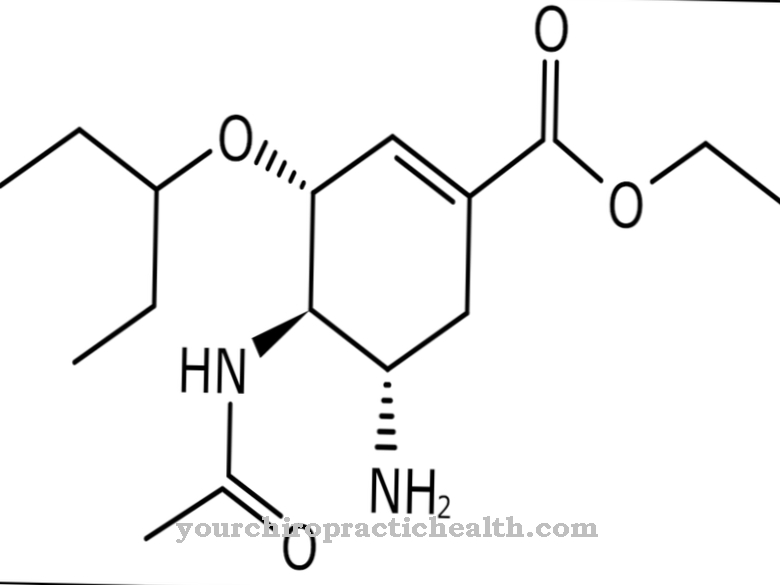
Estos fármacos se conocen por el tratamiento de la osteoporosis y tienen una gran afinidad por la superficie ósea. Especialmente en la región de las lagunas de reabsorción, se adhieren a los huesos. Al hacerlo, inhiben los osteoclastos que degradan los huesos y de esta manera reducen la resorción ósea. Los fármacos son análogos de pirofosfato con un enlace P-O-P que contiene carbono.
Sobre ellos no se produce una hidrólisis enzimática. Los aminobisfosfonatos se encuentran entre las más recientes de estas sustancias. Además, alendronato, clodronato, etidronato, ibandronato, pamidronato y risendronato están aprobados en Alemania del mismo grupo de fármacos. Lo mismo ocurre con el tiludronato y el zoledronato.
Además de estos medicamentos, los trasplantes de médula ósea también se pueden usar para tratar la enfermedad de células I. El éxito de este tratamiento solo ha sido limitado en casos anteriores. Las terapias genéticas ahora se están investigando como un nuevo enfoque terapéutico para los defectos genéticos. Las terapias genéticas han mostrado un éxito inicial en modelos animales. Hasta ahora, no se han podido utilizar en la práctica en seres humanos. Sin embargo, esta relación presumiblemente cambiará en el futuro.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el dolorOutlook y pronóstico
La enfermedad de células I es una enfermedad hereditaria que aún no se ha tratado sintomáticamente. Por tanto, el pronóstico es negativo. Aunque los síntomas pueden reducirse significativamente con una terapia temprana, la enfermedad de células I casi siempre toma un curso serio.
La baja estatura y el daño a los órganos internos y la cabeza ya reducen considerablemente la esperanza de vida. Además, las malformaciones en el rostro, la piel y los ojos pueden reducir la esperanza de vida, pero principalmente también la calidad de vida de la persona afectada. Algunos de los afectados llegan a los 40 o 50 años, pero la mayoría muere en la infancia o la adolescencia.
Si la enfermedad de las células I no se trata, los enfermos suelen morir en los primeros años de vida. Por tanto, el pronóstico es bastante negativo. Sin embargo, la perspectiva de una vida relativamente libre de síntomas se da si el paciente es tratado como parte de una terapia integral y, si es necesario, es internado en una institución para personas con discapacidad física. La fisioterapia y las medidas terapéuticas pueden mejorar significativamente el bienestar del paciente a largo plazo.
prevención
La enfermedad de células I solo se puede prevenir mediante una prueba genética molecular antes de la planificación familiar. Como parte del diagnóstico prenatal, los futuros padres también pueden decidir interrumpir el embarazo.
Cura postoperatoria
En la mayoría de los casos, las personas afectadas por la enfermedad de células I tienen muy pocas medidas de seguimiento disponibles o ninguna. La enfermedad debe ser reconocida por un médico lo antes posible para prevenir un empeoramiento adicional de los síntomas. Dado que se trata de una enfermedad determinada genéticamente, siempre se debe realizar un examen genético y una consulta en primer lugar en caso de deseo de tener hijos para evitar la herencia de la enfermedad de células I a la descendencia.
La mayoría de los pacientes dependen de varios medicamentos para esta enfermedad. Es importante asegurarse de que la dosis sea correcta y de que el medicamento se tome con regularidad. Si algo no está claro, hay efectos secundarios o si tiene alguna pregunta, siempre debe consultar primero a un médico.
Asimismo, muchos enfermos necesitan apoyo psicológico con esta enfermedad, aunque las conversaciones amorosas con los padres o familiares pueden tener un efecto positivo en el curso de la enfermedad. Una persona afectada necesita la ayuda y el apoyo en la vida diaria de su propia familia. En muchos casos, la enfermedad de células I limita o reduce significativamente la esperanza de vida de la persona afectada.
Puedes hacerlo tu mismo
Los pacientes que padecen la enfermedad de las células I pueden recurrir a varios métodos de tratamiento conservadores y alternativos. La terapia conservadora se enfoca en aliviar síntomas y dolencias.
El uso de ayudas como muletas o plantillas ortopédicas puede ralentizar el curso de las respectivas malformaciones y así también reducir el dolor. Los medicamentos ayudan a aliviar el dolor y se pueden complementar con medidas alternativas como masajes o acupuntura. Las terapias alternativas deben discutirse con el médico responsable de antemano. El médico puede derivar al paciente directamente a un homeópata o dar más consejos sobre cómo tratar el síntoma respectivo.
Dado que la enfermedad de células I suele ser mortal a pesar de todas las opciones de tratamiento, se debe buscar asesoramiento terapéutico. No solo los afectados deben superar sus miedos. Los familiares y amigos también suelen necesitar apoyo para afrontar la enfermedad y su posible resultado negativo. La participación en un grupo de autoayuda también es una opción para el paciente y sus familiares. El contacto con otros pacientes ayuda a aceptar la enfermedad y, a menudo, otros pacientes también pueden sugerir medidas de tratamiento y estrategias adicionales para la vida diaria con la enfermedad de células I.
.jpg)
.jpg)

.jpg)

.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)