En el Deficiencia de α-N-acetilgalactosaminidasa es una enfermedad de almacenamiento lisosomal que ocurre muy raramente. Se divide en forma juvenil y adulta.
¿Qué es la deficiencia de a-N-acetilgalactosaminidasa?

La deficiencia de a-N-acetilgalactosaminidasa o Deficiencia de alfa-N-acetilgalactosaminidasa también se llama en medicina Enfermedad de Schindler, Enfermedad de Schindler o Enfermedad de Kanzaki designado. Se trata de una enfermedad de almacenamiento lisosómico extremadamente rara, cuya herencia es autosómica recesiva. Los médicos diferencian entre una forma juvenil, que se presenta en niños y adolescentes, y una forma adulta, que sufren los adultos.
La forma juvenil de la deficiencia de alfa-N-acetilgalactosaminidasa se llama enfermedad de Schindler, mientras que la forma adulta se llama enfermedad de Kanzaki. Pero también hay subdivisiones en la enfermedad de Schindler tipo I para la forma juvenil y la enfermedad de Schindler tipo II para la forma adulta. También es posible una mezcla de ambas formas, conocida como enfermedad de Schindler tipo III.
Las tres formas de la enfermedad son oligosacaridosis (glicoproteinosas), que incluyen fucosidosis y enfermedad por almacenamiento de ácido siálico. El nombre de enfermedad de Schindler o enfermedad de Schindler para la deficiencia de alfa-N-acetilgalactosaminidasa se remonta al genetista alemán Detlev Schindler.
Fue el primero en describir la enfermedad en 1988. Un año después, el médico japonés T. Kanzaki describió la forma adulta. Descubrió en 1991 que una deficiencia de alfa-N-acetilgalactosaminidasa era la causa de la enfermedad.
causas
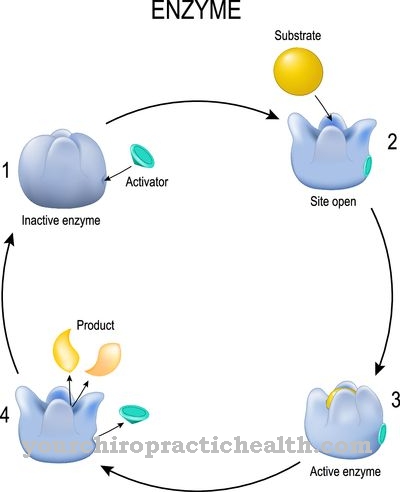
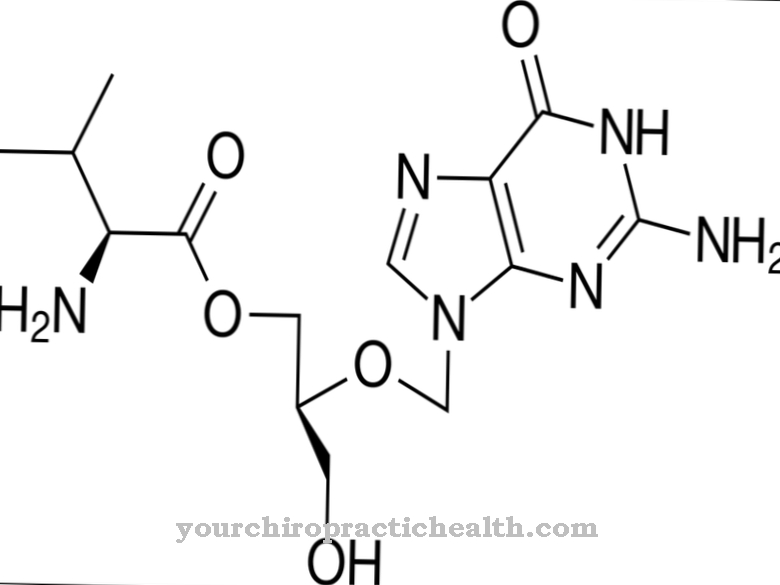
La deficiencia de α-N-acetilgalactosaminidasa es causada por una actividad reducida de la enzima alfa-N-acetilgalactosaminidasa. Las mutaciones sin sentido o sin sentido en el gen NAGA, que codifica la alfa-N-acetilgalactosaminidasa, son responsables del defecto enzimático.
El gen NAGA se encuentra en el locus q11 del gen del cromosoma 22. La enzima alfa-N-acetilgalactosaminidasa tiene la tarea de catalizar la escisión de N-acetilgalactosamina de varios glicolípidos y glicoproteínas. El defecto genético provoca una actividad reducida de la enzima, por lo que las sustancias que no se metabolizan se acumulan en las células del organismo.
En la mayoría de los casos se trata de proteoglicanos, glucoesfingolípidos y glucoproteínas ligadas a N u O. La acumulación de estos daña la célula y los órganos afectados. La deficiencia de alfa-N-acetilgalactosaminidasa es tan rara que no hay información precisa sobre su frecuencia.
Hasta ahora, se sabe que solo doce pacientes de ocho familias han desarrollado la enfermedad de Schindler en el mundo. Las quejas neurológicas no ocurren en todos los pacientes. Algunos médicos sospechan la influencia de factores adicionales para la aparición de síntomas neurológicos. Sin embargo, todavía no hay evidencia que respalde esta suposición.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el dolorSíntomas, dolencias y signos
Los síntomas de la deficiencia de alfa-N-acetilgalactosaminidasa dependen de la forma particular de la enfermedad. En el contexto de la enfermedad de Schindler tipo I, la hipotonía muscular progresiva ocurre en el primer año de vida. Además, los bebés afectados sufren una regresión psicomotora pronunciada.
También se registraron como síntomas alteraciones de la secuencia de movimiento, tetraplejía espástica y convulsiones cerebrales mioclónicas. También existe el riesgo de que el niño quede ciego. Si hay una forma adulta de deficiencia de alfa-N-acetilgalactosaminidasa, es decir, enfermedad de Schindler tipo 2, se presentan síntomas similares a la enfermedad de Fabry.
Los pacientes tienen angioqueratomas, que son cambios benignos de la piel. Muchos pacientes tienen un retraso mental leve. Una forma clínica intermedia es la enfermedad de Schindler tipo III, y los afectados padecen disfunciones neurológicas, convulsiones cerebrales y discapacidades mentales.
Sin embargo, también son posibles formas menos graves que se asocian con quejas psiquiátricas y neurológicas leves, retraso en el desarrollo del lenguaje o síntomas similares al autismo.
Diagnóstico y curso
El diagnóstico de una deficiencia de alfa-N-acetilgalactosaminidasa es posible mediante la detección de una actividad reducida de NAGA. Para ello, se realizan pruebas enzimáticas en plasma sanguíneo, leucocitos o fibroblastos o linfoblastos cultivados. La detección cromatográfica de oligosacáridos en la orina también permite el diagnóstico de deficiencia enzimática.
También se puede realizar un análisis de ADN, pero esto no es necesario en la mayoría de los casos. Durante el embarazo, el diagnóstico prenatal de la deficiencia enzimática también se puede realizar mediante un análisis de mutación del gen NAGA después de una muestra de vellosidades coriónicas o amniocentesis. Sin embargo, no se puede determinar la forma respectiva de la enfermedad.
También es importante el diagnóstico diferencial del síndrome de Fabry, la neurodegeneración asociada a pantotenato quinasa y la distrofia neuroaxonal infantil, que también tienen defectos enzimáticos. El curso de la deficiencia de alfa-N-acetilgalactosaminidasa depende de la forma respectiva de la enfermedad.
La enfermedad de Schindler tipo I se considera desfavorable, mientras que el pronóstico para los otros dos tipos es más favorable. Sin embargo, el número de personas enfermas es tan pequeño que no se pueden hacer declaraciones significativas sobre la esperanza de vida.
Complicaciones
Los síntomas y complicaciones de la deficiencia de A-N-acetilgalactosaminidasa dependen en gran medida de su forma.En la mayoría de los casos, sin embargo, ocurren trastornos del movimiento, por lo que los afectados están relativamente restringidos en su vida cotidiana. También ocurren calambres, que están asociados con un dolor intenso.
Los bebés, en particular, padecen graves trastornos del desarrollo, que pueden provocar espasticidad en el niño. En algunos casos, la deficiencia de A-N-acetilgalactosaminidasa conduce a una ceguera completa en el paciente. La regresión de la inteligencia conduce al retraso y, por tanto, a la discapacidad.
Suelen limitar considerablemente la vida diaria del paciente y repercutir negativamente en la calidad de vida. A menudo, los afectados dependen de la ayuda de otras personas. El retraso también puede conducir a un trastorno de búsqueda de palabras o un trastorno del lenguaje. No es posible tratar la deficiencia de A-N-acetilgalactosaminidasa de manera causal.
Por este motivo, la persona afectada debe tomar antibióticos. Ciertas complicaciones también deben evitarse directamente. La neumonía, por ejemplo, es uno de ellos. Además, el paciente también puede depender de la nutrición artificial.
¿Cuándo deberías ir al médico?
La deficiencia de A-N-acetilgalactosaminidasa definitivamente debe ser tratada por un médico. Esta deficiencia no desaparece por sí sola y no hay curación espontánea de esta enfermedad. En cualquier caso, se debe consultar a un médico si los padres notan una disminución de las capacidades motoras y mentales de su hijo o bebé. Las quejas con movimientos o procesos simples también pueden indicar la deficiencia de A-N-acetilgalactosaminidasa y deben ser examinadas por un médico.
Además, varias espasticidades también son un signo de enfermedad. No es raro que los pacientes sufran discapacidades intelectuales y otras disfunciones. Si estas quejas están presentes, definitivamente es necesario un tratamiento. Si no hay tratamiento, esto puede dar lugar a quejas y complicaciones considerables en la edad adulta y, por lo tanto, reducir considerablemente la calidad de vida de la persona afectada.
El desarrollo del habla muy retrasado también puede ser un signo de deficiencia de A-N-acetilgalactosaminidasa y, por lo tanto, debe investigarse. Como regla general, se puede consultar a un médico de cabecera para determinar la deficiencia de A-N-acetilgalactosaminidasa. A continuación, un especialista lleva a cabo el tratamiento adicional de las quejas individuales.
Doctores y terapeutas en su área
Tratamiento y Terapia
No es factible una terapia causal para la deficiencia de alfa-N-acetilgalactosaminidasa. Por lo tanto, el enfoque médico se limita a tratar los síntomas. Esto incluye una dieta sensata y una ingesta de líquidos, la prevención de enfermedades infecciosas, lo que también se puede hacer mediante la administración de antibióticos, así como frenar los ataques cerebrales mediante la administración de fármacos antiepilépticos.
Los pasos adicionales del tratamiento incluyen medidas de fisioterapia para prevenir la neumonía y las contracturas y para reducir el dolor o la espasticidad con la ayuda de medicamentos. Además, la profilaxis por aspiración puede realizarse mediante alimentación artificial.
Estudios recientes han demostrado que la deficiencia de a-N-acetilgalactosaminidasa es un trastorno de plegamiento de proteínas, por lo que se está discutiendo la terapia de reemplazo enzimático como una medida de tratamiento sensata. La terapia génica es otro enfoque terapéutico concebible para el tratamiento de trastornos de almacenamiento lisosómico. Dado que la enfermedad de Schindler se hereda de manera autosómica recesiva, se recomienda el asesoramiento genético de las familias afectadas.
Outlook y pronóstico
La deficiencia de A-N-acetilgalactosaminidasa puede provocar diferentes síntomas. En la mayoría de los casos, sin embargo, la deficiencia conduce a un retraso en el desarrollo psicomotor. En la edad adulta, el paciente a menudo depende de la ayuda de otras personas y no puede hacer frente a la vida cotidiana por sí solo. También ocurren trastornos del movimiento. Esto se puede notar por una cojera o por alteraciones de la coordinación.
En el peor de los casos, el paciente puede quedarse ciego o sufrir graves alteraciones visuales debido a la deficiencia de A-N-acetilgalactosaminidasa. No es raro que ocurran discapacidades intelectuales que reduzcan en gran medida la calidad de vida de la persona en cuestión. También ocurren trastornos del habla y trastornos del hallazgo de palabras, que pueden afectar la vida. Los niños en particular se ven afectados por el acoso y las burlas debido a los síntomas de la deficiencia de A-N-acetilgalactosaminidasa.
No es posible un tratamiento causal de la deficiencia de A-N-acetilgalactosaminidasa, por lo que el tratamiento está dirigido principalmente a reducir los síntomas. Se pueden utilizar varias medidas terapéuticas para reducir los trastornos del desarrollo. En muchos casos, sin embargo, el paciente depende de la ayuda externa en la vida diaria. La esperanza de vida no se ve afectada por la deficiencia.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el dolorprevención
No se conocen medidas preventivas contra la deficiencia de alfa-N-acetilgalactosaminidasa. La enfermedad de Schindler es una de las enfermedades extremadamente raras.
Cura postoperatoria
Dado que la deficiencia de A-N-acetilgalactosaminidasa es una enfermedad congénita que no se puede tratar de manera causal sino solo sintomática, el tratamiento completo no es posible. La persona afectada depende de un tratamiento de por vida, por lo que las opciones de cuidados posteriores son muy limitadas.
Como regla general, debido a la deficiencia de A-N-acetilgalactosaminidasa, el paciente suele depender de la ingesta de medicamentos y antibióticos. Deben tenerse en cuenta las posibles interacciones con otros medicamentos, aunque los antibióticos no deben tomarse junto con el alcohol. Si tiene un ataque, debe ir al hospital o llamar directamente a un médico de emergencia.
Además, los pulmones deben conservarse para evitar la inflamación. Por lo tanto, los pacientes con deficiencia de A-N-acetilgalactosaminidasa no deben fumar bajo ninguna circunstancia. Una dieta saludable y un estilo de vida generalmente saludable tienen un efecto muy positivo en el curso de la enfermedad.
Si la paciente desea tener hijos, el asesoramiento genético es útil para evitar que la enfermedad se transmita a los hijos. Dado que los pacientes a menudo sufren de movilidad restringida, la fisioterapia es útil para aliviar esto. También se pueden realizar muchos ejercicios en su propia casa.
Puedes hacerlo tu mismo
Una deficiencia de A-N-acetilgalactosaminidasa existente es una enfermedad incurable. Por tanto, el tratamiento médico es fundamental. Las medidas de autotratamiento solo pueden basarse en los síntomas para facilitar la vida diaria. Es importante que los padres observen al niño de cerca.
Los padres pueden ayudar mejor a sus hijos mediante la comprensión y la paciencia, el amor y el cuidado. Además, los tratamientos de terapia ocupacional y logopedia siempre se basan en un doble concepto: terapia presencial con el especialista y realización de los ejercicios en casa. Los padres deben ser activos aquí de manera constante y con paciencia. Se debe garantizar una dieta rica en vitaminas y minerales, ejercicio regular y mucho aire fresco. Esto fortalece el sistema inmunológico y reduce el riesgo de infección. Si la persona tiene tendencia a convulsionar, no debe estar sola durante mucho tiempo y debe ser examinada para ver si puede lesionarse si se produce una convulsión.
La mayoría de las veces, los pacientes no pueden hacer frente a la vida diaria por sí mismos y requieren cuidados constantes. Si los padres no pueden pagar esto, especialmente con niños mayores y adultos, no deben tener miedo de buscar ayuda. Esto puede tomar la forma de una enfermera o la colocación en una instalación adecuada. Si aún desea tener hijos, se recomienda una consulta con un especialista, ya que la enfermedad se hereda.
.jpg)
.jpg)




.jpg)



.jpg)