los Síndrome de Smith-Lemli-Opitz es un síndrome de malformación cogenital. La causa es una de un total de 70 mutaciones genéticas en el cromosoma 11q13.4. La enfermedad se hereda de forma autosómica recesiva y es una enfermedad extremadamente rara con múltiples malformaciones orgánicas y alteración de la biosíntesis del colesterol.
¿Qué es el síndrome de Smith-Lemli-Opitz?

© jijomathai - stock.adobe.com
los Síndrome de Smith-Lemli-Opitz entra en el grupo de síndromes de malformaciones hereditarias autosómicas recesivas. Una mutación genética provoca un trastorno metabólico en la biosíntesis del colesterol en la enfermedad. El síndrome es el trastorno cogenital más común de la biosíntesis de colesterol. La prevalencia de la enfermedad en Europa está entre 1: 60.000 y 1: 10.000.
Por tanto, la enfermedad puede clasificarse como una enfermedad rara, aunque es una de las enfermedades cogenitales más frecuentes en la biosíntesis del colesterol. El síndrome es aún más raro en los continentes de Asia y África. La enfermedad se describió por primera vez en 1964. Los genetistas D. W. Smith, L. Lemli y J. Marius Opitz documentaron el complejo de síntomas desde un punto de vista científico. Desde entonces se han notificado poco más de 300 casos.
Los niños se ven afectados con más frecuencia que las niñas. Es probable que los síntomas sean más pronunciados en las niñas y, por lo tanto, más difíciles de diagnosticar. La enfermedad es congénita, pero se desarrolla progresivamente desde el nacimiento y, por lo tanto, se caracteriza por diversas formas. El síndrome se divide en tipos I y II según los síntomas.
causas
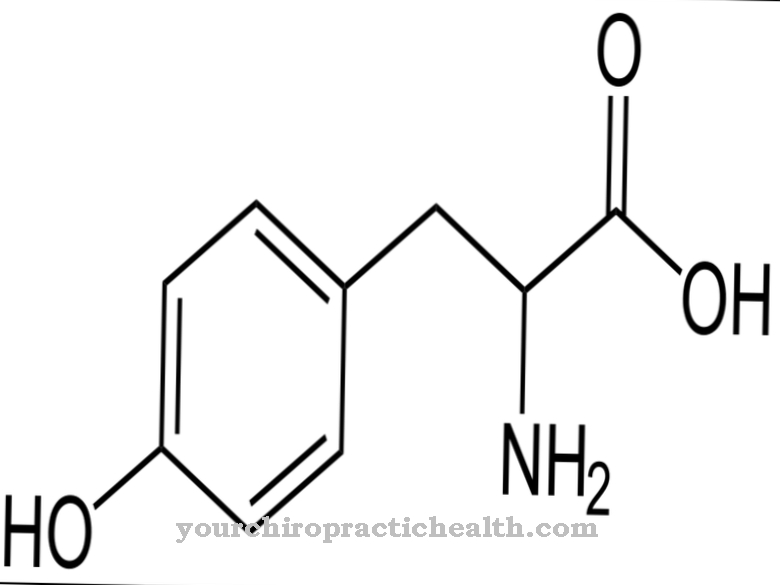
El síndrome de Smith-Lemli-Opitz es causado por una mutación genética que se localizó en 1998. El cromosoma 11q13.4 ahora se considera la ubicación de la mutación, con más de 70 mutaciones diferentes en esta ubicación conocidas hasta la fecha. El tipo de mutación causal determina la gravedad y el tipo de síntomas en cada caso individual. El gen en cuestión es el gen de la 7-esterol reductasa. S. Tint descubrió junto con sus colegas que el síndrome impide la producción de colesterol del propio cuerpo.
Esta producción incluye la conversión del precursor 7-dehidrocolesterol en el propio colesterol del cuerpo, que no puede funcionar debido a un defecto enzimático como resultado de la mutación. Por tanto, existe un exceso de 7-dehidrocolesterol en el organismo de los afectados. Al mismo tiempo, existe un déficit generalizado de colesterol. Debido a la herencia autosómica recesiva del síndrome, ambos padres deben ser portadores del gen defectuoso y solo pueden transmitirlo a un hijo de esta manera. Existe una probabilidad del 25 por ciento de que los siguientes hijos de padres con un hijo enfermo también se vean afectados por el síndrome de malformación.
Síntomas, dolencias y signos
Los niños con síndrome de Smith-Lemli-Opitz nacen con malformaciones craneofaciales típicas, especialmente microcefalia, una frente prominente y una nariz pequeña con una raíz nasal ancha. Además de las narinas antevertidas, hay microgenius. También se observan a menudo paladar hendido y opacidad del cristalino, especialmente cataratas y cataratas.
También está presente la blefaroptosis. Mental y cerebralmente, se desarrollan desarrollos indeseables con el tiempo, que resultan en una discapacidad mental. La holoprosencefalia y la irritabilidad también pueden moldear el cuadro clínico. Además del comportamiento de autolesión, el síndrome puede provocar un comportamiento autista. Además, existen múltiples malformaciones orgánicas, especialmente las del corazón y del tracto urogenital.
Las hipospadias y la criptorquidia son las malformaciones urogenitales más frecuentes. Además del exceso de dedos de manos o pies, puede haber sindactilia de los pies. La hipotonía muscular, los trastornos de la deglución y el reflujo gastroesofágico también son relevantes en el contexto del complejo de síntomas. También son frecuentes la dismotilidad intestinal y la estenosis pilórica. En el tipo II del síndrome existe pseudohermafroditismo, en el que los genitales externos son femeninos, aunque predomina el karotipo masculino.
Diagnóstico y curso de la enfermedad
Como diagnóstico prenatal, los exámenes de ultrasonido pueden registrar las características físicas típicas del síndrome de Smith-Lemli-Opitz incluso antes del nacimiento. Además de un déficit de crecimiento, se puede notar un defecto cardíaco o la ausencia de un riñón. En la prueba de líquido amniótico, es posible que el análisis de mutación ya proporcione un resultado que confirme el diagnóstico.
Después del nacimiento, los niños tienen una forma característica de la cara y posiciones especiales de las extremidades, por lo que el diagnóstico de sospecha se puede realizar mediante un diagnóstico visual si el diagnóstico prenatal ha fracasado. El diagnóstico genético asegura la sospecha. En términos de diagnóstico diferencial, el síndrome de Smith-Lemli-Opitz debe distinguirse del síndrome de alcoholismo fetal, el síndrome de Pallister Hall, el síndrome de Kaufmann-McKusick y el síndrome de Cornelia de Lange.
El síndrome de Pätau, el síndrome ATR-X y el síndrome C, el síndrome de Zellweger y el síndrome del hidroletalo también deben considerarse en el diagnóstico diferencial. Lo mismo ocurre con el Síndrome Oro-Faciales-Digitales, el Síndrome de Holoprosencefalia-Polidactilia y el Síndrome de Meckel. La esperanza de vida de los niños depende de la concentración de colesterol y de la tratabilidad de las malformaciones orgánicas. Los niveles bajos de colesterol y las malformaciones más graves hacen probable un desenlace fatal pronto. Los niños con niveles altos de colesterol y malformaciones fácilmente tratables no tienen una esperanza de vida gravemente afectada.
Complicaciones
Debido al síndrome de Smith-Lemli-Opitz, los afectados padecen diversas malformaciones y deformidades. Éstos tienen un efecto muy negativo sobre la calidad de vida del paciente. Los órganos internos en particular se ven afectados por las malformaciones, por lo que a menudo la muerte puede ocurrir inmediatamente después del nacimiento. Además, la mayoría de los pacientes padecen paladar hendido y también problemas oculares.
Además, este síndrome a menudo conduce a discapacidades intelectuales y, por lo tanto, a retraso intelectual. Por lo tanto, la mayoría de los pacientes dependen de la ayuda de otras personas en su vida y ya no pueden hacer frente a muchas tareas diarias por sí mismos. El corazón también se ve afectado por las malformaciones, que pueden provocar una muerte cardíaca súbita. Además, el síndrome de Smith-Lemli-Opitz también afecta a los genitales, por lo que también pueden producirse malformaciones en estos.
El tratamiento del síndrome de Smith-Lemli-Opitz generalmente solo puede ser sintomático. No hay complicaciones y algunos de los síntomas pueden ser limitados. Sin embargo, no se puede lograr un curso completamente positivo de la enfermedad. No se puede predecir universalmente si la esperanza de vida será limitada.
Tratamiento y Terapia
Para los pacientes con síndrome de Smith-Lemli-Opitz, la atención médica y social de por vida es a menudo inevitable. Por regla general, su desarrollo está muy retrasado en las áreas cognitiva y motora. En casi todos los casos, esto resulta en una discapacidad de por vida que no permite un estilo de vida independiente. Por eso, sobre todo, se proporciona un tratamiento de apoyo.
Como parte de estas medidas, los padres reciben apoyo psicoterapéutico e idealmente aprenden a lidiar con la enfermedad de sus hijos. El síndrome de Smith-Lemli-Opitz es incurable y, por lo tanto, no puede tratarse como una causa. Dado que se ha documentado un trastorno del metabolismo del colesterol para el síndrome, es posible un tratamiento sintomático para compensar la deficiencia de colesterol. Este tratamiento se realiza administrando colesterol.
Las malformaciones, en su mayoría numerosas de los órganos, deben corregirse quirúrgicamente, en la medida de lo posible. Una excepción a esto es el enlace múltiple frecuentemente documentado de los dedos de las manos y los pies, que no requiere necesariamente una intervención quirúrgica. Los síntomas concomitantes, como las dificultades visuales, ahora también se pueden tratar y aliviar bien.
La mayoría de los afectados padecen problemas nutricionales como dificultades para succionar y tragar o reflujo gastroesofágico y alteración del peristaltismo gastrointestinal. Por lo tanto, a menudo se debe usar una sonda gástrica para garantizar una ingesta segura de alimentos. Los problemas de conducta posiblemente se puedan tratar con terapia conductual.
prevención
Después de un diagnóstico prenatal positivo del síndrome de Smith-Lemli-Opitz, los padres tienen la oportunidad de interrumpir el embarazo. El síndrome de Smith-Lemli-Opitz solo se puede prevenir de otra manera si las parejas tienen un diagnóstico genético molecular realizado en su planificación familiar y, una vez que se han proporcionado pruebas sobre la mutación, deciden en contra de sus propios hijos.
Cura postoperatoria
Las medidas de seguimiento para el síndrome de Smith-Lemli-Opitz (SLOS) se basan en la gravedad de los síntomas que aparecen en el curso de la enfermedad. En la mayoría de los casos de enfermedad, los niños tienen problemas nutricionales. Lo hacen mal. Por lo tanto, el enfoque de la atención de seguimiento es, en primer lugar, la nutrición adecuada de los niños afectados mediante alimentos líquidos nasogástricos ricos en calorías y la administración de colesterol suficiente.
El curso posterior de la enfermedad también muestra en muchos de los niños afectados un subdesarrollo del cerebro. Este subdesarrollo generalmente conduce a discapacidades físicas o mentales con diversos grados de gravedad. Por ejemplo, no todos los niños afectados aprenden a caminar. Para compensar la movilidad restringida, se proporcionan ayudas para el movimiento diario (por ejemplo, silla de ruedas, ayudas para caminar y pararse) como medida de seguimiento en este caso.
En caso de síntomas mentales como autoagresión e hiperactividad, se continúa con el tratamiento farmacológico prescrito terapéuticamente como medida de seguimiento. Además, alrededor del 50 por ciento de todos los niños afectados desarrollarán un defecto cardíaco de moderado a grave a medida que avanza la enfermedad. Después de la operación del defecto cardíaco, se programan exámenes electrocardiográficos (EKG) y ecográficos a intervalos regulares.
Para los padres de niños con SLOS, se recomiendan consultas y terapias psicológicas. La vida independiente en la edad adulta es bastante improbable. Es de esperar que se tomen medidas de cuidados intensivos en la atención de seguimiento del SLOS en la edad adulta. Además, las malformaciones de órganos pueden limitar la esperanza de vida.
Puedes hacerlo tu mismo
La enfermedad se asocia con numerosas quejas que perjudican gravemente la calidad de vida. Si a un miembro de la familia se le ha diagnosticado la enfermedad genética, se debe consultar a un médico antes de concebir al niño. Se deben sopesar los posibles riesgos para que se puedan tomar decisiones prudentes para todos los involucrados. Además, debe participar en todos los controles que se ofrecen durante el embarazo.
Tan pronto como se noten los problemas de salud del niño, los padres y familiares pueden tomar las precauciones adecuadas y prepararse mejor para futuros desarrollos. En una gran cantidad de casos, cuidar a alguien con síndrome de Smith-Lemli-Opitz es un desafío enorme para los familiares. Por lo tanto, deben conocer y respetar sus límites físicos y emocionales. Es recomendable considerar el uso de la atención médica para el paciente y, en paralelo, el apoyo psicoterapéutico para los familiares. Como resultado, a menudo se pueden lograr mejoras en el tratamiento de la enfermedad.
Cabe señalar que la estabilidad del entorno social es importante para el paciente. Además, siempre debe mantener la calma en la vida cotidiana cuando surjan adversidades y desafíos. Dado que la persona en cuestión no está en condiciones de moldear su vida de forma independiente, debe haber una empatía especial al tratar con él.

.jpg)
.jpg)

.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)