los Enfermedad por almacenamiento de ácido siálico es una enfermedad de almacenamiento lisosomal genético muy poco frecuente en la que la proteína sialina está codificada incorrectamente. La sialina es una proteína transmembrana que normalmente elimina los monosacáridos como el ácido siálico, que surgen de la descomposición enzimática de las glicoproteínas y otras sustancias, del lisosoma. La funcionalidad reducida de la sialina conduce a una acumulación de ácido siálico en los lisosomas, es decir, a una enfermedad por almacenamiento de ácido siálico.
¿Qué es la enfermedad por almacenamiento de ácido siálico?

© LIGHTFIELD STUDIOS - stock.adobe.com
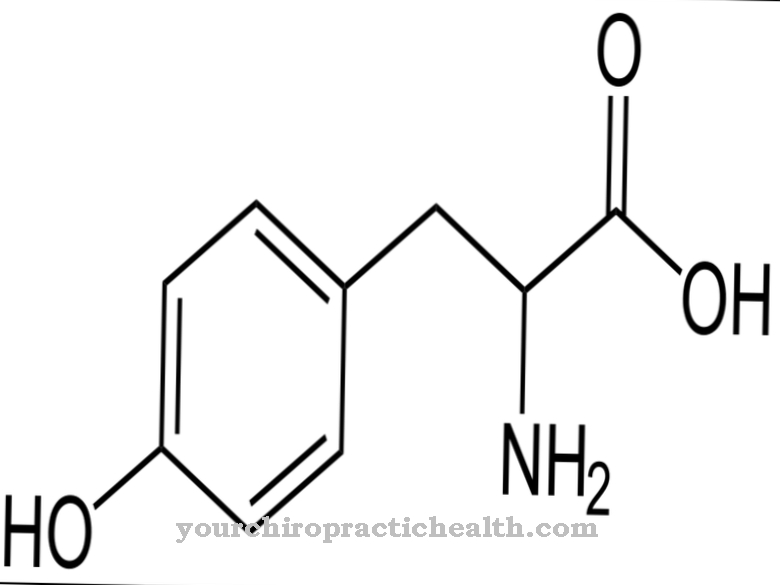
El término ácido siálico incluye derivados de ácido neuramínico que están N- u O-acetilados. Los ácidos siálicos son componentes básicos de glicoproteínas, glicosaminoglicanos y otras sustancias y se liberan a través de su descomposición enzimática dentro de lisosomas especiales.
Normalmente, los ácidos siálicos se canalizan fuera del lisosoma a través de la membrana del lisosoma con la ayuda de la proteína transportadora de aniones sialina. Característica de la Enfermedad por almacenamiento de ácido siálico, que también se llaman Enfermedad por almacenamiento de ácido neuramínico es una restricción funcional o una pérdida total de función de la proteína transportadora sialina.
Los ácidos siálicos producidos en los lisosomas se pueden canalizar fuera de los lisosomas, por lo que hay una acumulación excesiva de ácidos siálicos en los lisosomas. Se conocen una forma infantil y una forma adulta de enfermedad por almacenamiento de ácido siálico lisosómico. La forma adulta también se conoce como enfermedad de Salla.
causas
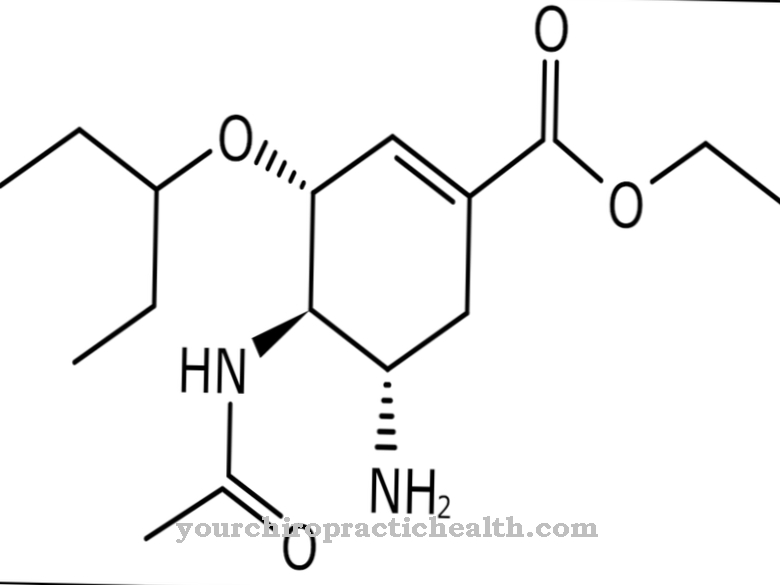
Las causas de la pérdida de función de la proteína transmembrana sialina se remontan a un defecto o una mutación del gen SLC17A5 tanto en la forma infantil como en la adulta. El gen está en el cromosoma 6, en el locus del gen q14-q15. Todas las mutaciones conocidas del gen conducen a una codificación incorrecta de la proteína sialina.
Aunque el metabolismo lo acepta como un componente básico, no tiene la capacidad de descargar ácido siálico a través de la membrana del lisosoma. La codificación incorrecta de la sialina le da a la proteína una estructura terciaria diferente, que está asociada con la pérdida de funcionalidad como proteína de transporte. Diferentes mutaciones genéticas conducen a la forma infantil o adulta de la enfermedad por almacenamiento de ácido siálico.
Síntomas, dolencias y signos
La enfermedad por almacenamiento de ácido siálico infantil es la que tiene un pronóstico menos favorable de ambas formas. Los primeros signos y síntomas se manifiestan antes del nacimiento. El diagnóstico prenatal por ultrasonido generalmente revela la llamada hidropesía fetal, una acumulación de líquido tisular que se extiende por grandes partes del feto.
Después del parto, el agrandamiento del hígado y el bazo es sintomático y los rasgos faciales son en su mayoría ásperos. La esperanza de vida en la forma infantil de la enfermedad por almacenamiento de ácido neuramínico suele ser de unos pocos años. La enfermedad de Salla o la forma adulta de la enfermedad por almacenamiento de ácido siálico ya se nota en la infancia.
Esto sucede por hipotensión muscular y un nistagmo horizontal, en el que los ojos realizan movimientos horizontales espasmódicos. Los afectados presentan discapacidades mentales y en su mayoría no tienen capacidad de lenguaje. El pronóstico de la esperanza de vida es ligeramente más favorable que el de la forma infantil de la enfermedad. Los afectados suelen llegar a la edad adulta.
Diagnóstico y curso de la enfermedad
En las personas afectadas por la enfermedad por almacenamiento de ácido siálico, una mayor concentración de ácido siálico en la orina y una mayor tasa de excreción de polisacáridos son sintomáticos. El ácido siálico libre se puede detectar en fibroblastos y células amnióticas. Una prueba genética, que también se puede realizar de forma prenatal, sirve como certeza final.
En casos graves de enfermedad por almacenamiento de ácido siálico, las malformaciones óseas, la ascitis (ascitis), los trastornos graves del movimiento, los calambres espásticos y las discapacidades mentales graves se notan poco después del nacimiento. Las formas graves de la enfermedad provocan la muerte en la primera infancia.
Con cursos moderados, como son típicos de la enfermedad de Salla, los afectados suelen llegar a la edad adulta. Las discapacidades intelectuales graves, las convulsiones espásticas y las restricciones psicomotoras graves son síntomas de estas formas de enfermedad. Aunque la enfermedad es muy rara en todo el mundo, tiene un enfoque regional en el norte de Finlandia, donde cada 40 portadores heterocigotos es una de las mutaciones genéticas correspondientes.
Complicaciones
En la mayoría de los casos, la enfermedad por almacenamiento de ácido siálico se puede reconocer y diagnosticar antes del nacimiento. Los afectados suelen sufrir un agrandamiento significativo del bazo y del hígado. Esto puede provocar dolor en las respectivas regiones. Los rasgos faciales de los pacientes suelen ser toscos, por lo que pueden producirse burlas o acoso, especialmente a una edad temprana.
Muchos niños sufren problemas psicológicos o depresión. Además, los afectados a menudo padecen discapacidad intelectual y, por tanto, también restricciones en la vida cotidiana. Por lo tanto, a menudo dependen de la ayuda de otras personas en su vida cotidiana y no pueden hacer fácilmente muchas cosas cotidianas. La enfermedad por almacenamiento de ácido siálico también puede provocar problemas de lenguaje, por lo que la comunicación entre los afectados se restringe considerablemente.
Debido a la enfermedad por almacenamiento de ácido siálico, la esperanza de vida del paciente también suele reducirse considerablemente. La mayoría de los afectados solo llegan a la edad adulta con él. Dado que la terapia causal no es posible, solo se pueden limitar los síntomas. Sin embargo, no hay complicaciones particulares. La enfermedad por almacenamiento de ácido siálico no se puede prevenir.
¿Cuándo deberías ir al médico?
La enfermedad por almacenamiento de ácido siálico generalmente siempre requiere examen y tratamiento médicos. Dado que se trata de una enfermedad hereditaria, también conviene realizar una prueba genética si quieres tener hijos para evitar que la enfermedad se transmita a tus descendientes. Dado que esto no conduce a la autocuración, la persona afectada por la enfermedad por almacenamiento de ácido siálico depende en cualquier caso del tratamiento de por vida de un médico.
Se debe consultar a un médico si el paciente tiene un bazo o hígado significativamente agrandados. En la mayoría de los casos, estos síntomas se descubren por casualidad mediante un examen de ultrasonido, aunque el dolor en los lados del cuerpo también puede indicar estos síntomas. En algunos casos, los problemas para hablar o una serie de discapacidades intelectuales apuntan a una enfermedad por almacenamiento de ácido siálico.
El primer examen de esta denuncia puede realizarlo un médico de cabecera. El tratamiento adicional generalmente lo llevan a cabo varios especialistas y siempre se basa en los síntomas exactos.
Tratamiento y Terapia
Dado que la enfermedad por almacenamiento de ácido siálico se basa en un defecto genético, no es posible una terapia causal. No hay forma de reemplazar la proteína transmembrana y de transporte de aniones funcionalmente deteriorada con sialina funcional. Las posibles terapias se limitan (todavía) a tratar los síntomas y garantizar que el paciente experimente el menor dolor posible.
En el caso de algunas otras enfermedades por almacenamiento lisosómico, como la enfermedad de Hunter y la enfermedad de Gaucher, la infusión de enzimas modificadas genéticamente ya ha mostrado resultados positivos. El procedimiento se conoce como terapia de reemplazo enzimático y es eficaz en los órganos internos y el tejido conectivo. Debido a su tamaño, las enzimas suministradas artificialmente no pueden atravesar la barrera hematoencefálica, por lo que tales terapias no influyen en el desarrollo del SNC.
Una forma de terapia que todavía está en su infancia es la llamada terapia génica, que tiene como objetivo conducir a un intercambio dirigido de la secuencia del gen mutado. En principio, se trata de una técnica que se utiliza en sentido positivo en la reparación de genes del propio organismo o también puede tener efectos negativos en forma de determinados gérmenes patógenos.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el dolorprevención
No existen medidas preventivas que puedan prevenir el brote de la enfermedad por almacenamiento de ácido siálico, porque es una enfermedad sistémicamente eficaz, determinada genéticamente, que ya se ha creado de forma prenatal.
En familias en las que, por ejemplo, se han conocido casos de enfermedad de Salla, si desean tener hijos, se puede realizar un examen genético para determinar si el padre potencial es portador de una de las mutaciones genéticas conocidas sin estar enfermo. En el caso positivo, debe realizarse una consulta en la que se expliquen los riesgos que existen para el niño en la realización del deseo de tener hijos.
Cura postoperatoria
Como regla general, los pacientes con enfermedad por almacenamiento de ácido siálico no tienen opciones especiales para la atención de seguimiento directa. Por esta razón, esta enfermedad debe ser diagnosticada y tratada temprano por un médico para que no haya complicaciones u otros síntomas en el curso posterior.
Cuanto antes se consulte a un médico, mejor será el curso posterior de esta enfermedad, por lo que se debe contactar a un médico tan pronto como aparezcan los primeros signos y síntomas. La enfermedad por almacenamiento de ácido siálico no suele curarse por sí sola. La mayoría de los pacientes con enfermedad por almacenamiento de ácido siálico dependen de tomar varios medicamentos para aliviar los síntomas de la enfermedad de forma permanente y adecuada.
Se debe observar el consumo regular y la dosis prescrita para contrarrestar correctamente los síntomas. Los chequeos regulares por parte de un médico también son muy importantes para identificar y tratar otros daños en una etapa temprana. Debido a la enfermedad, algunos de los afectados también dependen de la ayuda y el apoyo de sus propias familias. Esta enfermedad también puede reducir la esperanza de vida del paciente.
Puedes hacerlo tu mismo
Dado que la enfermedad se basa en un defecto genético, la persona afectada no puede tomar ninguna opción que contribuya a la cura del trastorno de salud.El enfoque de las medidas de autoayuda debe estar dirigido a mejorar los procesos diarios y el bienestar.
Hay que tener en cuenta que una gran cantidad de pacientes padecen un deterioro del rendimiento cognitivo. Por lo tanto, a menudo dependen de la ayuda y el apoyo de familiares o personal de enfermería con formación médica. A la hora de afrontar la enfermedad, los padres u otros miembros de la familia en particular deben asegurarse de tener en cuenta sus propios límites y no entregarse a la atención y el apoyo. El desarrollo de elementos para mejorar la alegría de vivir es de inmensa importancia para todos los involucrados. Un entorno social estable también ayuda con la adversidad.
Dado que la enfermedad tiene un efecto negativo en el desarrollo de algunos órganos, esto debe tenerse en cuenta en la vida cotidiana. Una dieta saludable, una higiene óptima del sueño y una rutina diaria regular ayudan a hacer frente a la enfermedad. Aportan seguridad y estabilidad. Dado que la comunicación del paciente es limitada, se deben desarrollar posibilidades para que se produzca un buen intercambio de todos los involucrados en la vida cotidiana. La comprensión mutua, la calma y evitar el ritmo frenético son beneficiosos para un mayor desarrollo.

.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)