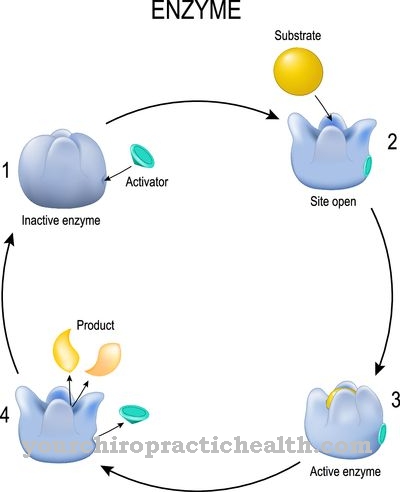
los Lipoproteína lipasa (LPL) pertenece a las lipasas y juega un papel crucial en el metabolismo de los lípidos. Es responsable de dividir los triglicéridos en los quilomicrones y las lipoproteínas de muy baja densidad (VLDL) en ácidos grasos y monoacilglicerina. Los ácidos grasos liberados se utilizan para generar energía o para acumular grasa corporal.
¿Qué es la lipoproteína lipasa?
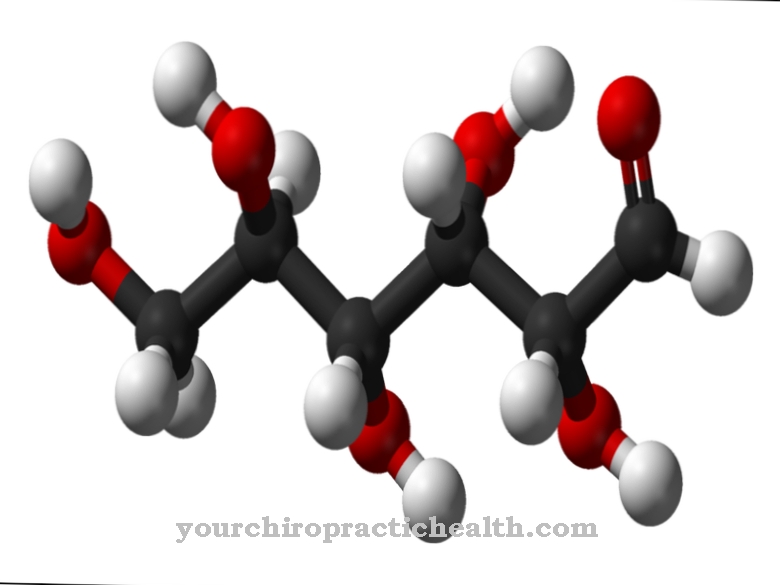
La lipoproteína lipasa (LPL) es una enzima que es una de las lipasas. Las lipasas son responsables de descomponer los triglicéridos (triacilgliceroles) en ácidos grasos y glicerina. Los triglicéridos son ésteres de la glicerina de triple alcohol con tres ácidos grasos cada uno y se conocen como grasas o aceites grasos.
Las grasas de la dieta se absorben con los alimentos y primero son degradadas por las lipasas extracelulares del páncreas en el intestino. Sin embargo, algunos triglicéridos ingresan al torrente sanguíneo a través del suero cuando se absorben en el intestino delgado, donde se unen a lipoproteínas que garantizan su capacidad de ser transportados en la sangre. La lipoproteína lipasa es la enzima que descompone los triglicéridos unidos a las lipoproteínas en ácidos grasos y monoacilglicerol. Consta de 448 aminoácidos y depende de la coenzima apolipoproteína C2 para su función.
La lipoproteína lipasa es una enzima soluble en agua que se une a las células endoteliales de los vasos sanguíneos a través de ciertas glicoproteínas (proteoglicanos). Se produce en el hígado. La enzima cataliza la hidrólisis de los triglicéridos para formar dos moléculas de ácidos grasos y una molécula de monoacilglicerol. Las apolipoproteínas son las moléculas portadoras de las triglicerinas y les permiten ser transportadas en un ambiente acuoso. La apolipoproteína C2 también actúa como receptor de la lipoproteína lipasa y, por tanto, activa la hidrólisis de los triglicéridos.
Función, efecto y tareas
La función de la lipoproteína lipasa es catalizar completamente la descomposición en la sangre de las grasas absorbidas por las células intestinales. Primero, las grasas de la dieta se descomponen en ácidos grasos y glicerina por las lipasas pancreáticas en el intestino delgado. Más triglicéridos ingresan a la sangre a través de la absorción a través del intestino delgado y allí se unen a las lipoproteínas para formar un complejo lípido-proteína.
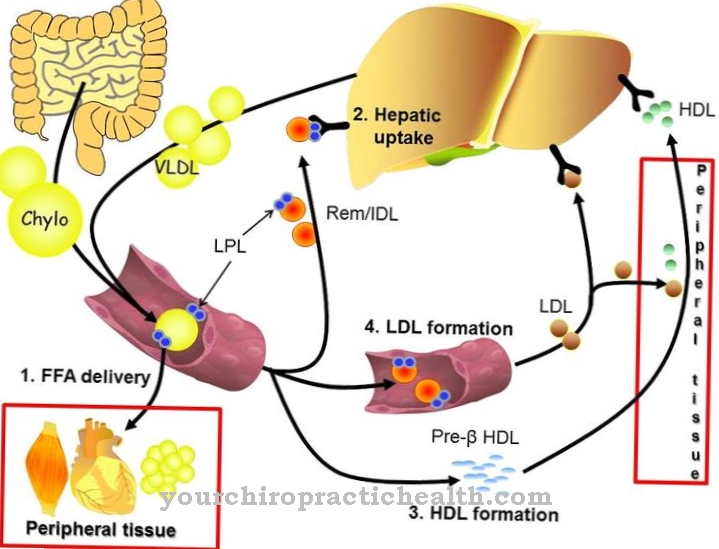
Esto crea quilomicrones. Representan partículas de lipoproteínas con un diámetro de 0,5 a 1 micrómetro y su densidad es inferior a 1000 g / ml. El núcleo lipídico contiene principalmente triglicéridos con una pequeña cantidad de ésteres de colesterol. La capa que contiene colesterol de los quilomicrones contiene fosfolípidos como elemento estructural. Las apolipoproteínas a las que se unen los triglicéridos ahora también se almacenan en esta capa. Los quilomicrones contienen 90 por ciento de triglicéridos. Entran en el torrente sanguíneo desde el intestino delgado a través del sistema linfático. Los triglicéridos se descomponen en ácidos grasos y glicerina con la ayuda del LPL, especialmente en los capilares del tejido muscular y adiposo.
Los ácidos grasos se usan en el tejido muscular para generar energía o en el tejido graso para acumular triglicéridos endógenos como grasa de almacenamiento. Después de aproximadamente diez horas de abstinencia de alimentos, no se pueden detectar más quilomicrones en la sangre porque los triglicéridos se descomponen por completo. Otros componentes de la sangre son las llamadas VLDL (lipoproteínas de muy baja densidad). Estas unidades estructurales se liberan del hígado y contienen triglicéridos, fosfolípidos y colesterol. Las VLDL transportan estos componentes a través del torrente sanguíneo desde el hígado hasta los órganos individuales.
De esta manera, los triglicéridos son degradados por la lipoproteína lipasa y los ácidos grasos liberados son absorbidos por las células del cuerpo. La disminución de los triglicéridos convierte la VLDL en LDL (lipoproteína de baja densidad). Las LDL contienen principalmente fosfolípidos, ésteres de colesterol y lipoproteínas.
Educación, ocurrencia, propiedades y valores óptimos
La lipoproteína lipasa se sintetiza en el hígado. Además de las lipasas pancreáticas, representa otra lipasa extracelular La LPL se encuentra en el exterior de las membranas de las células endoteliales de varios órganos, incluidas las células grasas. Allí se conecta a las membranas celulares a través de los llamados proteoglicanos.
Sin embargo, es de particular importancia para las células endoteliales de los vasos sanguíneos, ya que aquí puede controlar directamente la hidrólisis de los triglicéridos en los quilomicrones y VLDL. Se inyecta heparina para medir la actividad de las lipoproteasas. La heparina elimina la unión de las lipoproteínas lipasas de los proteoglicanos, de modo que después de una inyección de heparina hay una mayor concentración de lipoproteínas lipasas libres, que puede determinarse por su actividad. Este examen puede, entre otras cosas, determinar una deficiencia de lipoproteína lipasa.
Enfermedades y trastornos
La falta de lipoproteína lipasa a menudo conduce a graves problemas de salud. Si hay muy poca lipoproteína lipasa o si su actividad es insuficiente debido a un defecto genético, los triglicéridos en los quilomicrones y las VLDL solo se pueden descomponer mal o no descomponerse.
La deficiencia de lipoproteína lipasa puede ser principalmente genética, así como secundaria a la quimioterapia, por ejemplo. La deficiencia primaria de LPL es rara y está causada por un defecto genético autosómico recesivo. Se desarrolla la denominada quilomicronemia, que se caracteriza por un suero lechoso y cremoso y se denomina hiperlipidemia tipo I. Los triglicéridos en los quilomicrones ya no se descomponen. Como resultado, se producen una y otra vez pancreatidas graves con intolerancia a la leche y dolor abdominal.
Además, se desarrollan constantemente xantomas explosivos y hepatomegalia. Las únicas opciones de tratamiento son una dieta baja en grasas y sin alcohol. Esta enfermedad a menudo es causada por mutaciones en el gen LPL en el cromosoma 8 o en el gen APOC2. La forma secundaria de hiperlipidemia tipo I generalmente ocurre con la quimioterapia y es solo de naturaleza temporal.





.jpg)



.jpg)