los Síndrome de Kearns-Sayre (KSS) se describió sistemáticamente por primera vez en 1958 y es una de las enfermedades mitocondriales muy raras y determinadas genéticamente. los KSS tiene una sintomatología central con pocos síntomas que se presenta en todos los pacientes. A lo largo de la vida hay otras enfermedades graves, según los tejidos afectados por los defectos mitocondriales. Deben tratarse por separado.
¿Qué es el síndrome de Kearns-Sayre?

© L.Darin - stock.adobe.com
los Síndrome de Kearns-Sayre afecta aproximadamente a 12 de cada 100.000 pacientes. La enfermedad muy rara es genética y se manifiesta en una alteración del metabolismo intracelular. El ADN de las mitocondrias en las células está dañado por mutación en un total de 4977 pares de bases. Eso corresponde a 12 genes mitocondriales.
Estas células defectuosas se encuentran en los músculos esqueléticos, los músculos externos del ojo y en las células del hígado. Dado que las mitocondrias mutadas ya no pueden convertir (correctamente) los nutrientes proporcionados por la ingesta de alimentos en energía para las células, gran parte de los músculos y el tejido circundante ya no reciben trifosfato de adenosina (ATP).
A diferencia de otras enfermedades de las células musculares, las que progresan lentamente KSS Otros tejidos también se ven afectados: Dependiendo de qué órganos ya no reciben ATP adecuadamente, daño al músculo cardíaco, debilidad muscular, pérdida auditiva, diabetes mellitus, retraso en la pubertad, problemas gastrointestinales. Los primeros síntomas generalmente se diagnostican a los 10 años. Solo hay varios cientos de casos documentados de la enfermedad en todo el mundo.
causas
La muy rara enfermedad genética ocurre solo de forma esporádica, por lo que la medicina asume que se trata de mutaciones espontáneas. Ambos sexos se ven afectados por igual. Las mutaciones mitocondriales se transmiten de madre a hijo. Los médicos hablan de una herencia autosómica recesiva en relación con la herencia del síndrome de Kearns-Sayre.
Síntomas, dolencias y signos
Los síntomas básicos del lubricante refrescante aparecen alrededor de los 10 años. El paciente tiene párpados caídos (ptosis). También existen trastornos del movimiento ocular (oftalmoplagia externa progresiva, CPEO). El trastorno del pigmento de la retina (retinopatía pigmentosa atípica) provoca alteraciones visuales y una restricción del campo visual. Más adelante, puede provocar una atrofia muscular progresiva con alteración de la coordinación del movimiento (ataxia). Los reflejos se reducen (hiporreflexia) o ya no ocurren en absoluto (arreflexia).
Si el músculo cardíaco está dañado, el resultado son trastornos de conducción (arritmias cardíacas). El deterioro de las células nerviosas conduce a trastornos sensoriales en las extremidades. Además, los pacientes tienen una audición reducida. El deterioro mental hasta la demencia también puede ocurrir en el contexto del síndrome de Kearns-Sayre.
El equilibrio hormonal alterado conduce a la diabetes mellitus, una tiroides hipoactiva y un trastorno del crecimiento (estatura baja). Algunos pacientes también desarrollan trastornos del lenguaje y la deglución.
Diagnóstico y curso de la enfermedad
Los principales síntomas del síndrome de Kearns-Sayre se reconocen desde el principio, y la mayoría de los demás aparecen más tarde a medida que la enfermedad progresa lentamente. El análisis de LCR muestra un contenido de proteínas de más de 100 mg / dl en pacientes con SK. El nivel en sangre muestra niveles elevados de lactato y piruvato. Los cambios estructurales mitocondriales se hacen visibles bajo el microscopio electrónico. La TC muestra la calcificación de los ganglios basales.
Con la ayuda de la EMG, se puede demostrar una disminución de la actividad muscular. Los trastornos de conducción de estímulos se pueden determinar con la ayuda del ENG. Una biopsia de los músculos esqueléticos muestra un resultado positivo cuando se altera de 1,3 a 10 kb de ADN mitocondrial y se pueden identificar las típicas "fibras rojas desgarradas". El electrorretinograma hace visible la "retina de sal y pimienta".
La miocardiopatía (latidos cardíacos irregulares) se diagnostica mediante un ECHO y un EKG. Sin embargo, el médico tratado solo puede estar 100% seguro de que el paciente examinado tiene síndrome de Kearns-Sayre si ha examinado todo el ADN mitocondrial con la ayuda de amplificación por PCR y ha realizado un análisis de secuencia de la secuencia de ADNmt destruida.
Complicaciones
El síndrome de Kearns-Sayre conlleva considerables restricciones en la vida cotidiana y en la vida de la persona afectada. En la mayoría de los casos, los pacientes padecen trastornos visuales graves y una limitación del campo visual. Además, puede producirse ceguera completa en el curso posterior. No es raro que el paciente experimente problemas cardíacos, que pueden provocar la muerte cardíaca.
La sensibilidad de la persona afectada suele ser limitada y se produce parálisis. La mayoría de los pacientes también padecen demencia u otros trastornos mentales, por lo que a menudo tienen que depender de la ayuda de otras personas en su vida diaria. Esto también puede provocar trastornos del habla.
La coordinación del movimiento también se ve afectada, lo que conduce a restricciones de movimiento u otros trastornos de la marcha. La calidad de vida disminuye significativamente debido al síndrome de Kearns-Sayre. En muchos casos, los padres o familiares de los afectados también necesitan tratamiento psicológico para prevenir la depresión u otras dolencias psicológicas.
El tratamiento se lleva a cabo con la ayuda de medicamentos. En muchos casos, es necesario un trasplante de corazón, ya que generalmente puede provocar una muerte cardíaca súbita. La esperanza de vida del paciente está severamente limitada por el síndrome de Kearns-Sayre.
¿Cuándo deberías ir al médico?
Los padres deben consultar a un médico con sus hijos si hay cambios visuales en los párpados. En caso de enfermedad, estos cuelgan repentinamente y no se pueden cambiar mediante una tensión muscular intencional. Los signos del síndrome de Kearns-Sayre se desarrollan a la edad de diez años y requieren evaluación y tratamiento médicos. Los trastornos del movimiento ocular, las deficiencias visuales y una reacción refleja reducida se consideran inusuales y deben aclararse. En la mayoría de los casos, el campo de visión de la persona afectada es limitado. La audición reducida, los trastornos cardíacos o las interrupciones del ritmo cardíaco deben ser examinados por un médico lo antes posible.
Si el niño sufre de sensibilidad anormal en las extremidades, necesita ayuda médica. El adormecimiento de la piel o la hipersensibilidad a la irritación deben presentarse a un médico. Si se trata de trastornos de la memoria, irregularidades en la memoria o rendimiento mental reducido, es necesaria una visita al médico.
Los exámenes médicos son aconsejables en caso de baja estatura, problemas de conducta y trastornos del lenguaje. Si el niño se queja de dificultad para tragar, se niega a comer o si pierde peso, se debe consultar a un médico. Si hay una ingesta insuficiente de líquidos, el organismo se ve amenazado por un suministro insuficiente. Se debe consultar a un médico para que no se desencadene una afección potencialmente mortal. Los pacientes con síndrome de Kearns-Sayre necesitan un médico tan pronto como los síntomas aumentan.
Terapia y tratamiento
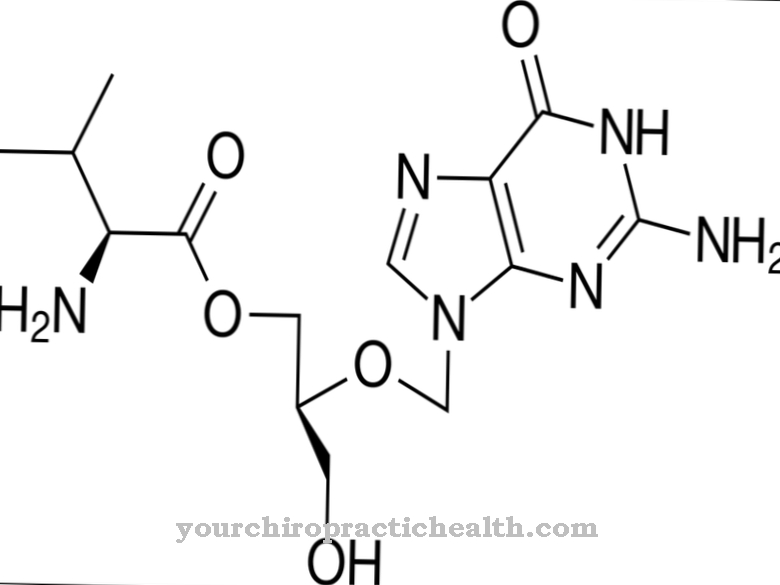
La cura para el síndrome de Kearns-Sayre aún no es posible. Los pacientes generalmente tienen una esperanza de vida más corta, pero esta se puede prolongar con las medidas médicas adecuadas. En casos extremos, la miocardiopatía puede provocar una muerte cardíaca súbita. No existe una terapia causal. Sin embargo, en la mayoría de los pacientes con lubricantes refrescantes, la coenzima Q10 (ubiquinona) altamente concentrada puede mejorar su condición. Por lo general, se consume una dosis de 30 a 260 mg por vía oral.
Alternativamente, se pueden tomar de 90 a 270 mg / día de idebenona. Los agentes mejoran la función mitocondrial en el cerebro y los músculos esqueléticos. Para otros defectos mitocondriales, también se prescriben carnitina o creatina, por ejemplo. De lo contrario, el tratamiento solo se puede administrar sintomáticamente. Las visitas regulares al cardiólogo ayudan en el tratamiento de las arritmias cardíacas crónicas. En algunos casos, se debe utilizar un marcapasos.
Si la afección es particularmente grave, incluso puede estar indicado un trasplante de corazón. La pérdida de audición se puede compensar al menos durante un cierto período de tiempo con un audífono adecuado. Las visitas oftalmológicas periódicas reducen el riesgo de complicaciones oculares. Si el síndrome de Kearns-Sayre está presente, el pronóstico depende de la gravedad de la enfermedad, es decir, de cuántos órganos están involucrados en el proceso de la enfermedad y de la proporción de ADNmt anormal presente en cada uno de ellos.
Outlook y pronóstico
Aunque el síndrome de Kearns-Sayre es poco común, el pronóstico del trastorno es malo. La causa es un defecto genético que no puede ni debe tratarse. Por razones legales, los científicos y los profesionales médicos no pueden cambiar la genética humana. Esto no puede curar el trastorno. Se lleva a cabo un tratamiento sintomático, que solo puede aliviar los síntomas existentes.
El paciente está expuesto a una terapia a largo plazo, que se asocia con diversas tensiones y efectos secundarios. Tan pronto como el tratamiento aplicado se interrumpa o finalice de forma independiente, es de esperar una recaída de las quejas. Muchas de las quejas existentes no pueden tratarse a pesar de todos los esfuerzos y opciones médicas.
En el caso de deficiencias mentales y trastornos del cerebro, no suele haber enfoques terapéuticos para mejorar la salud. En estos casos, se acompaña al paciente con los mejores medios posibles y se le proporciona una enfermera. Por lo general, no es posible afrontar la vida cotidiana. Debido a la gran cantidad de trastornos que desencadena el síndrome, puede suponer una fuerte carga emocional para la persona afectada y sus familiares. Los trastornos psicológicos pueden estallar y conducir a un mayor deterioro de la situación general. Esto debe tenerse en cuenta al realizar una previsión.
prevención
La prevención no es posible con el síndrome de Kearns-Sayre porque es una enfermedad genética. La génesis exacta aún no se ha aclarado con claridad. Sin embargo, es posible, con la ayuda de métodos especializados adecuados, reconocer los nuevos síntomas de la enfermedad crónica grave lo suficientemente temprano y tratarlos a tiempo.
Cura postoperatoria
En la mayoría de los casos, los afectados por el síndrome de Kearns-Sayre no tienen opciones de seguimiento especiales o directas disponibles, por lo que el paciente depende definitivamente de un diagnóstico rápido y, sobre todo, precoz de la enfermedad. Tan pronto como aparezcan los primeros signos y síntomas, la persona afectada debe, por tanto, consultar a un médico para evitar que los síntomas empeoren.
Dado que el síndrome de Kearns-Sayre es una enfermedad genética, se deben realizar pruebas genéticas y asesoramiento si desea tener hijos para evitar que la enfermedad recurra. La mayoría de las personas afectadas por el síndrome de Kearns-Sayre dependen de los medicamentos.
Siempre es importante tomarlo con regularidad y utilizar la dosis correcta para que los síntomas puedan aliviarse de forma permanente. También se debe visitar a un cardiólogo con regularidad para que se pueda controlar permanentemente el estado del corazón. En el peor de los casos, esto puede provocar una muerte súbita cardíaca, por lo que la esperanza de vida se ve reducida en muchos casos por el síndrome. El curso posterior de esta enfermedad depende en gran medida del momento del diagnóstico, por lo que no se puede hacer una predicción general al respecto.
Puedes hacerlo tu mismo
En el caso del síndrome de Kearns-Sayre, la medida de autoayuda más importante es adaptar de forma óptima la medicación a las condiciones de vida. Dado que los afectados tienen un mayor riesgo de muerte súbita cardíaca, también es importante estar atento a síntomas y quejas inusuales.
Si se notan dolores de corazón, dificultad para respirar, dolor y otros signos de la enfermedad, se debe alertar al médico de emergencia de inmediato. En casos menos graves, los pacientes solo necesitan tomarse las cosas con calma. Se deben evitar las actividades físicas agotadoras, así como el estrés y estimulantes como el alcohol, la cafeína o la nicotina. La pérdida de audición se puede compensar con un audífono adecuado. La vista se puede estabilizar al menos mediante visitas regulares al oftalmólogo.
Si su salud es particularmente mala, necesitará un trasplante de corazón. En preparación para este procedimiento, el paciente debe cambiar su dieta e informar al médico que está tomando algún medicamento. Las enfermedades, alergias o dolencias que el médico aún no conoce también deben aclararse antes del procedimiento. Después de la operación, el paciente necesita reposo y reposo en cama, acompañado de una atención médica cercana. Las otras medidas que pueda tomar la persona en cuestión dependerán de los órganos involucrados en el proceso de la enfermedad.
.jpg)





.jpg)



.jpg)