los Síndrome de Aicardi-Goutières es una mutación genética hereditaria autosómica recesiva extremadamente rara que desencadena un trastorno del desarrollo genéticamente heterogéneo en el cerebro de bebés de aproximadamente tres meses y, además de las deficiencias motoras graves, se asocia con espasticidad y epilepsia. Para los fenotipos severos, una expectativa de vida máxima de diez años es una guía, aunque los fenotipos más leves con mutaciones de RNASEH2B posiblemente pueden vivir más de diez años.
¿Qué es el síndrome de Aicardi-Goutières?

El médico entiende que el síndrome de Aicardi-Goutières es una enfermedad hereditaria autosómica recesiva que corresponde a un trastorno del desarrollo genéticamente heterogéneo del cerebro en bebés alrededor de los tres meses de edad. Clínicamente, el síndrome tiene un aspecto similar a una infección intrauterina, es decir, una infección que se adquirió en el útero, pero no se pueden detectar patógenos ya que existe una causa genética.
Ciertas enzimas del núcleo celular son menos activas de lo habitual debido al defecto genético y, por lo tanto, limpian los cromosomas significativamente menos de proteínas de ARN falso. Esto conduce a una acumulación de segmentos de ADN en las células. Como esto hace que las células mueran, el sistema inmunológico se activa y causa inflamación en las áreas relevantes.
En 1984, los médicos franceses Françoise Goutières y Jean François Aicardi describieron por primera vez este fenómeno. Desde entonces, se han conocido menos de 150 casos, por lo que el defecto genético es igualmente raro. Hasta ahora, los casos individuales se han observado principalmente en familias, ya que un segundo hijo también desarrollará el síndrome con una probabilidad del 25 por ciento.
causas
No existe una forma adquirida de síndrome de Aicardi-Goutières. El trastorno del desarrollo del cerebro es siempre congénito y se remonta a una mutación genética autosómica recesiva. Para desencadenar un brote de enfermedad, el gen defectuoso debe estar presente en ambos lados de la herencia. Sin embargo, también son posibles nuevas mutaciones. Hasta ahora, cinco ubicaciones de genes se han relacionado con la mutación.
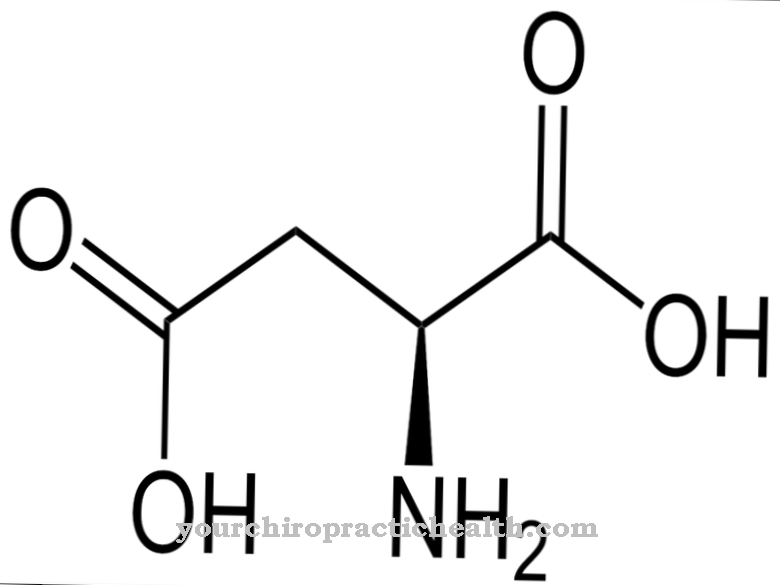
Estos incluyen TREX1, RNASEH2A, RNASEH2B y RNASEH2C, así como SAMHD1. Las primeras descripciones del síndrome asumen una causa en relación con el gen TREX1 en el cromosoma 3p21. Los genes RNASEH2-A son loci de genes para tres proteínas diferentes que desempeñan un papel en la formación de la enzima del núcleo celular trimérico RNasa H.
Esta enzima del núcleo celular es a su vez responsable de eliminar los bloques de construcción de moléculas de ARN colocados incorrectamente del ADN. Sin la RNasa H, se produce la muerte embrionaria temprana. Si, por otro lado, la enzima del núcleo celular está presente pero no está completamente activa, los fragmentos individuales de ciertos genes se almacenan en las células del cuerpo. Causan apoptosis, que desencadena un proceso inflamatorio basado en la reacción inmune natural.
Puedes encontrar tu medicación aquí
➔ Medicamentos para los calambres muscularesSíntomas, dolencias y signos
Los niños con síndrome de Aicardi-Goutières son difíciles de alimentar. Tiene movimientos oculares espasmódicos y está inquieto y sufre leves ataques de fiebre y vómitos. Un poco menos de la mitad de los niños afectados pierden las habilidades motoras que habían aprendido hasta ahora después de unos seis meses. En cuanto a la motricidad, aparecen en particular los signos piramidales.
Además de la parálisis espástica, puede haber movimientos descoordinados que hacen que los músculos de los brazos y las piernas se contraigan o provoquen convulsiones. Además de la alteración del tono muscular, a veces se agrandan órganos como el hígado y el bazo. Posteriormente hay un retraso creciente en el desarrollo psicomotor. Ocasionalmente, también ocurren cambios en la piel similares a la congelación.
Diagnóstico y curso
En el diagnóstico diferencial, el médico debe descartar principalmente los parásitos de Toxoplasma si se sospecha el síndrome de Aicardi-Goutières. La tomografía computarizada muestra calcificaciones en los ganglios basales a ambos lados del AGS. La pérdida de materia cerebral y las malformaciones de la materia cerebral también pueden ser indicaciones de la tomografía computarizada.
El examen del agua del nervio muestra un aumento del nivel de linfocitosis en el LCR e interferones alfa, que indican un proceso inflamatorio en el sistema nervioso central. En el suero de pacientes con AGS, las plaquetas sanguíneas se reducen y al mismo tiempo aumentan los valores de las enzimas hepáticas. En lo que respecta al curso de la enfermedad, muchos de los afectados todavía mueren en la infancia, siendo diez años la esperanza de vida máxima para las formas particularmente graves de la mutación.
Los pacientes con formas más leves a menudo viven hasta los diez años. Estos fenotipos más leves corresponden principalmente a mutaciones de RNASEH2B, que están presentes en casi la mitad de todos los casos. La enfermedad progresa al principio durante varios meses hasta que se presenta una discapacidad múltiple. Sin embargo, después de estos primeros meses, la enfermedad generalmente se estabiliza de manera constante. Aunque hasta ahora casi todos los pacientes mayores de diez años han tenido que luchar con discapacidades múltiples graves, en casos individuales se han producido cursos significativamente más leves.
Complicaciones
El síndrome de Aicardi-Goutières tiene graves complicaciones que pueden tener un impacto muy negativo en las personas y los niños. En la mayoría de los casos, el síndrome se manifiesta con dificultad para alimentarse y movimientos oculares espasmódicos y descontrolados. A medida que avanza la enfermedad, la fiebre y los vómitos se vuelven más comunes.
Con el síndrome de Aicardi-Goutières, los niños presentan parálisis espástica de brazos y piernas, por lo que ya no pueden coordinarse y moverlos correctamente. En el peor de los casos, pueden ocurrir convulsiones, que pueden ser particularmente peligrosas para los niños. El síndrome de Aicardi-Goutières también conduce a retraso mental.
En la mayoría de los casos, los síntomas conducen a la muerte en la infancia, por lo que la esperanza de vida desciende enormemente. Si el niño no muere con el síndrome de Aicardi-Goutières, habrá complicaciones y dificultades considerables en la vida cotidiana. Debido a las limitaciones físicas y psicológicas, el paciente siempre depende de la ayuda de otras personas y cuidadores.
Generalmente es necesaria una estadía en el hospital. El síndrome de Aicardi-Goutières también puede afectar el hígado y el bazo, provocando su agrandamiento. Esto suele provocar dolor en los órganos respectivos.
¿Cuándo deberías ir al médico?
En la mayoría de los casos, el síndrome de Aicardi-Goutières no puede ser tratado por un médico. El paciente muere después de los primeros diez años de vida. Como regla general, los síntomas ya los determina un pediatra. Sin embargo, los padres pueden comunicarse con un pediatra si hay retrasos notables en el desarrollo o en el desarrollo de varios tipos de espasticidad.
Es necesaria una visita al médico incluso con movimientos espasmódicos y fiebre fuerte y persistente. Por lo general, los medicamentos pueden limitar la fiebre y los vómitos. No es raro que los niños sufran parálisis y calambres. Estos también pueden estar restringidos por el tratamiento, por lo que en estos casos también se recomienda una visita al médico.
También se puede llamar a un médico de urgencias en caso de crisis o convulsiones agudas. No es raro que los padres sufran problemas psicológicos o depresión. En este caso, se recomienda el tratamiento psicológico de los padres. Esto debe recibir apoyo psicológico, especialmente después de que el niño muere por el síndrome de Aicardi-Goutières.
Doctores y terapeutas en su área
Tratamiento y Terapia
El síndrome de Aicardi-Goutières aún no se puede tratar de manera causal, solo se trata sintomáticamente. En este contexto, las convulsiones se pueden prevenir mediante la administración de fármacos antiepilépticos, por ejemplo. Se suelen utilizar medidas fisioterapéuticas para reducir la espasticidad y las contracturas.
Ciertos ungüentos y cremas se pueden usar contra los síntomas de la piel. Las opciones terapéuticas también incluyen la intervención temprana, en la que no solo se atiende a los niños afectados sino, sobre todo, a los padres.
Outlook y pronóstico
El síndrome de Aicardi-Goutières conduce a diversas malformaciones y tensiones para el paciente. Los padres del niño, en particular, a menudo se ven afectados por graves quejas psicológicas y depresión y, por lo tanto, necesitan apoyo psicológico.
El niño mismo sufre de graves discapacidades y ataques epilépticos. Esto provoca fiebre intensa y vómitos. A menudo, el niño también sufre de movimientos incontrolables de los ojos y parálisis en diferentes partes del cuerpo. Además, pueden producirse convulsiones, que a menudo se asocian con dolor. El desarrollo del niño está severamente restringido por el síndrome de Aicardi-Goutières y ocurren trastornos motores. El síndrome también afecta negativamente al sistema nervioso central. La calidad de vida del paciente es extremadamente limitada y la persona afectada suele depender de la ayuda de otras personas en la vida diaria.
El síndrome de Aicardi-Goutières no se puede tratar, por lo que solo se pueden limitar los síntomas. Los ataques epilépticos se pueden reducir relativamente bien con la ayuda de medicamentos. El paciente también dispone de varias terapias.
Puedes encontrar tu medicación aquí
➔ Medicamentos para los calambres muscularesprevención
Un feto puede ser examinado para el síndrome de Aicardi-Goutières mientras aún está en el útero tomando una muestra de líquido amniótico. Los padres que esperan entonces pueden decidir en contra del niño. No se dispone de otras medidas preventivas porque el síndrome es una mutación.
Cura postoperatoria
Las opciones de atención de seguimiento son relativamente limitadas en el síndrome de Aicardi-Goutières. El paciente siempre depende del tratamiento médico para paliar los síntomas y evitar posibles complicaciones. Por regla general, el síndrome también reduce considerablemente la esperanza de vida de la persona afectada.
Dado que la afección es hereditaria, no se puede tratar por completo. El paciente dispone de un tratamiento puramente sintomático. Si quieres tener hijos, también se puede realizar un asesoramiento genético para prevenir la recurrencia del síndrome de Aicardi-Goutières. Los afectados dependen en su mayoría de la medicación.
Estos deben tomarse con regularidad y también deben considerarse las posibles interacciones con otros medicamentos. Los niños reciben la terapia y el apoyo adecuados para contrarrestar la espasticidad. Sin embargo, no se pueden curar por completo.
El contacto con otras personas que padecen el síndrome de Aicardi-Goutières a menudo tiene un efecto positivo en el curso posterior de la enfermedad. No es raro que se intercambie información y se facilite la vida cotidiana. Además, los padres a menudo también dependen del tratamiento psicológico. Las conversaciones con amigos o conocidos también pueden resultar útiles.
Puedes hacerlo tu mismo
El síndrome de Aicardi-Goutières es una enfermedad con múltiples discapacidades. Los padres deberán buscar tratamiento médico permanente con su hijo. La carga para los padres es muy alta y se recomienda encarecidamente el apoyo psicológico. También se debe buscar sin dudarlo más ayuda profesional para el cuidado diario del niño.
Si hay otros niños en la familia, también debe discutirse la posibilidad de colocarlos en un centro de cuidado. Dado que el niño enfermo necesita cuidados intensivos, la carga no debe subestimarse. Para los padres con un hijo enfermo, el riesgo de depresión es particularmente mayor. No se debe descuidar el cuidado de la propia salud, física y mental. Los padres solo pueden ser un apoyo estable para sus hijos si ellos mismos son estables.
Las convulsiones y los vómitos frecuentes requieren una observación constante del niño. El niño necesita cuidados de sus padres o profesionales para todas las actividades de la vida diaria.
Los padres deben asegurar un movimiento suficiente mediante ejercicios motores, hacer que la dieta del niño sea rica en sustancias vitales y fácilmente digerible, y asegurar una ingesta adecuada de líquidos. El apoyo fisioterapéutico es particularmente importante para reducir los ataques espásticos y acompaña a la terapia con fármacos antiepilépticos. Los padres pueden tratar las dermatosis de forma solidaria aplicando cuidadosamente cremas y ungüentos.

.jpg)
.jpg)
.jpg)




.jpg)

.jpg)