Del Enfermedad de Pompe es una enfermedad por almacenamiento de glucógeno de curso impredecible. Los síntomas se caracterizan por una debilidad muscular progresiva. Mientras tanto, en terapia, se han observado éxitos mediante la administración artificial de la enzima causante.
¿Qué es la enfermedad de Pompe?

© designua - stock.adobe.com
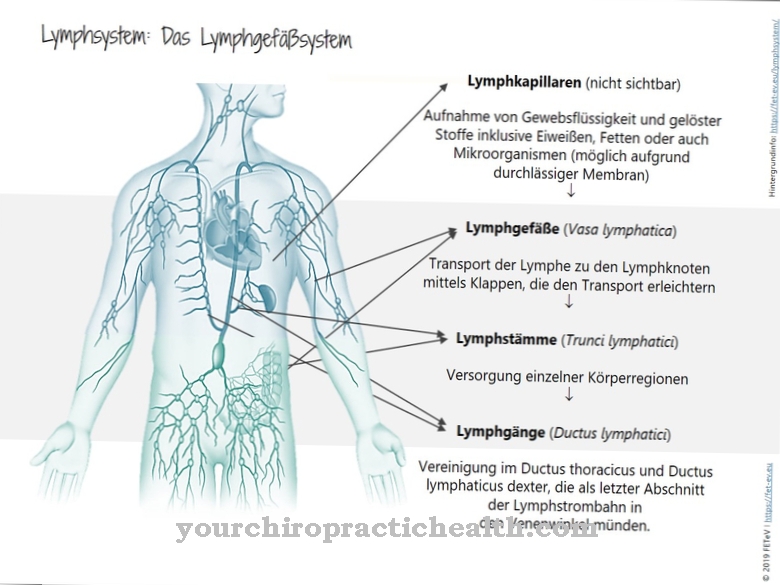
Las enfermedades por almacenamiento son un grupo heterogéneo de enfermedades en las que se depositan diversas sustancias en órganos o células del organismo. Los tejidos u órganos suelen perder su función debido a los depósitos. Las sustancias depositadas pueden ser de varios tipos. Dependiendo de la sustancia, se distingue entre glucogenosis, mucopolisacaridosis, lipidosis, esfingolipidosis, hemosiderosis y amiloidosis. En la glucogenosis, el glucógeno se almacena alrededor del tejido corporal.
Los carbohidratos almacenados ya no se pueden descomponer en absoluto o solo de manera incompleta o se pueden transformar en glucosa. La causa suele ser una deficiencia de enzimas que participan en la conversión de glucosa en el cuerpo. Una enfermedad por almacenamiento de glucógeno es que Enfermedad de Pompequien tambien Enfermedad de Pompian o Deficiencia de maltasa ácida se llama. La enfermedad afecta a la α-glucosidasa lisosomal o la maltasa ácida en el gen GAA.
En el cuerpo sano, la enzima descompone los polisacáridos de cadena larga de los lisosomas en glucosa. En los seres humanos, la enzima está presente en todos los tejidos. La enfermedad metabólica todavía se nota principalmente en los músculos y, por lo tanto, también se incluye en las miopatías. La rara enfermedad lleva el nombre del patólogo holandés Pompe, quien describió el fenómeno por primera vez en 1932. En 1954 G.T. La enfermedad de Cori como enfermedad de almacenamiento de glucógeno tipo II.
causas
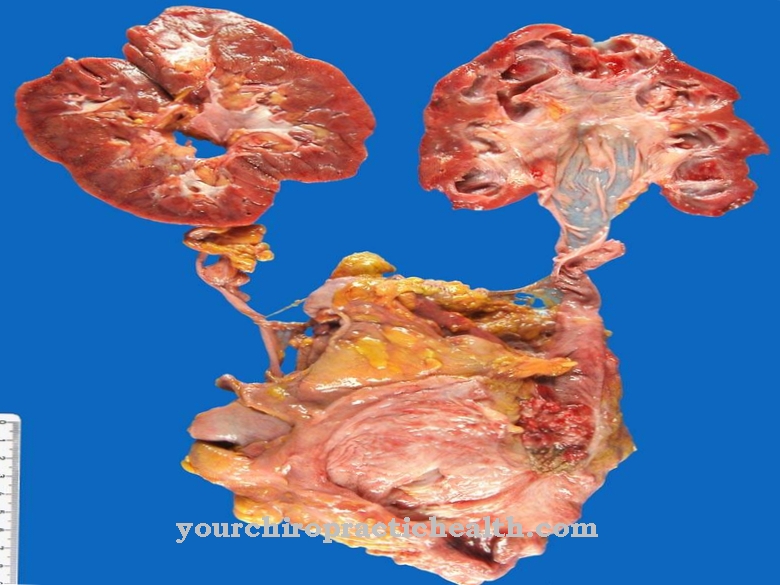
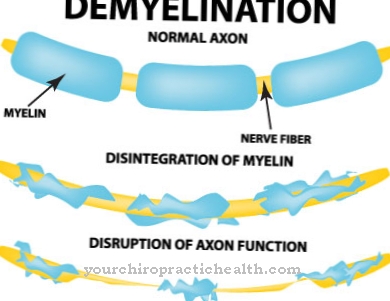
En la década de 1960, H.G. La suya es la falta de α-glucosidasa lisosomal como vínculo causal en la enfermedad de Pompe. Esta deficiencia surge sobre la base de un defecto genético principalmente causal que afecta a la enzima α-1,4-glucosidasa. Esta enzima también se conoce como maltasa ácida y está completamente ausente o tiene una actividad disminuida. Esto evita la degradación del glucógeno del almacenamiento de azúcar en los músculos. Por tanto, el glucógeno se almacena en las células musculares de los lisosomas, donde destruye las células musculares. La actividad restante de la enzima se correlaciona con la gravedad de la enfermedad.
El tipo infantil de enfermedad de Pompe tiene una actividad de menos del uno por ciento. El tipo juvenil retiene una actividad residual de hasta el diez por ciento y el tipo adulto retiene una actividad residual de hasta el 40 por ciento. La enfermedad está sujeta a herencia autosómica recesiva. Los niños se ven afectados con tanta frecuencia como las niñas. El defecto genético causal se localizó en la región q25.2-q25.3 del cromosoma 17 y tiene una longitud de 28 kpb.
La enfermedad es genéticamente heterogénea y hasta ahora se ha asociado con 150 mutaciones diferentes. Los pacientes son heterocigotos compuestos. La forma infantil se caracteriza a menudo por dos mutaciones graves. Con esta forma, existe un alto grado de correspondencia entre el genotipo y el curso de la enfermedad. Este no es el caso de la forma adulta. La prevalencia se da con valores entre 1: 40.000 y 1: 150.000. Alrededor de 200 personas han sido diagnosticadas en Alemania.
Síntomas, dolencias y signos
Los principales síntomas de la enfermedad de Pompe son cardiomegalia e insuficiencia cardíaca. También existen déficits neurológicos y musculares. El brote de la enfermedad de Pompe no se limita a una determinada edad, sino que puede afectar a todos los grupos de edad. La forma infantil se presenta en lactantes y generalmente termina fatalmente en el primer año de vida. En la mayoría de los casos, la muerte que se produce es la muerte por insuficiencia cardíaca, que se debe a una cardiomegalia hipertrófica.
En la forma infantil, los primeros síntomas aparecen a los dos meses. Los adolescentes desarrollan la enfermedad de Pompe juvenil. En los adultos, la medicina habla de la enfermedad de Pompe del adulto. Estas formas se manifiestan sintomáticamente en debilidad muscular progresiva en los músculos respiratorios y los músculos esqueléticos del tronco. La parte superior de los brazos puede verse afectada, así como la pelvis y los muslos. El curso es impredecible en las formas adulta y juvenil. Los cursos difíciles se caracterizan por una pérdida de transpirabilidad.
También suele haber una pérdida de movilidad. Surgen estados de agotamiento. En algunos casos, las sustancias se depositan en las arterias y pueden formar un aneurisma, cuya ruptura puede ser fatal. En promedio, los primeros síntomas se notan poco antes de los 30 años y corresponden a dificultades para correr o hacer ejercicio. El diagnóstico se suele realizar a mediados de los 30. Aproximadamente diez años después, los afectados dependen en su mayoría de una silla de ruedas.
Diagnóstico y curso de la enfermedad
El diagnóstico de enfermedad de Pompe generalmente se confirma mediante una biopsia muscular basada en la anamnesis. Histológicamente, los depósitos masivos de glucógeno en el músculo pueden demostrarse en la tinción PAS. El diagnóstico se puede anclar fácilmente en una medición de la actividad enzimática de la maltasa ácida, como se puede detectar en los leucocitos con el análisis de sangre seca. Los exámenes genéticos moleculares también pueden usarse como medio de diagnóstico. CK, CKMB, LDH, GOT y GPT aumentan en la sangre. La orina suele estar elevada en Glc4.
Deben descartarse numerosos diagnósticos diferenciales. En la forma infantil, debido a los síntomas pronunciados, el diagnóstico sospechoso a menudo se puede asegurar mediante la rápida progresión que se asocia con el aumento de los problemas respiratorios y el retraso del desarrollo motor. La cardiomegalia se puede confirmar mediante rayos X. En teoría, también es concebible un diagnóstico prenatal basado en el examen del líquido amniótico o la extracción de tejido.
El curso de la enfermedad de Pompe suele ser más grave cuanto antes estalla la enfermedad. No obstante, la enfermedad de Pompe se caracteriza por un curso individual y, por lo tanto, realmente impredecible de la enfermedad. También se observan formas leves.
Complicaciones
Con la enfermedad de Pompe, los afectados sufren importantes restricciones y quejas en la vida cotidiana. En primer lugar, existen dificultades respiratorias, que conducen a una gran reducción de la resiliencia y fatiga del paciente. Además, la falta de oxígeno también puede conducir a una pérdida del conocimiento, en la que la persona afectada puede resultar lesionada por una caída.
Las dificultades respiratorias también tienen un efecto negativo sobre el corazón y los demás órganos internos, por lo que pueden producirse daños consecuentes irreversibles en los órganos.La esperanza de vida se reduce y restringe significativamente por la enfermedad de Pompe. En el peor de los casos, la persona afectada puede morir de muerte cardíaca. Las actividades agotadoras o deportivas aún no son posibles para la persona afectada por esta enfermedad.
El tratamiento de la enfermedad de Pompe generalmente se basa en los síntomas y tiene como objetivo aumentar la esperanza de vida. Se utilizan diversos fármacos, que normalmente no conducen a complicaciones particulares. Los síntomas también se pueden reducir con la ayuda de fisioterapia. Si existen limitaciones psicológicas, es necesario un tratamiento psicológico adicional.
¿Cuándo deberías ir al médico?
Cualquiera que padezca la enfermedad de Pompe se ve afectado por la enfermedad hereditaria cuando era niño o tiene síntomas musculares graves en la edad adulta. Las personas con glucogenosis tipo II sufren una pérdida de masa muscular cada vez mayor. Los músculos respiratorios también pueden verse afectados. Si el defecto genético no se descubre temprano en un examen de rutina, las visitas al médico están a la orden del día a medida que aumentan los síntomas.
Sin embargo, el diagnóstico puede llevar mucho tiempo. Primero, hay muchas enfermedades con un curso similar. En segundo lugar, las pruebas genéticas no son la norma en medicina. En tercer lugar, la enfermedad por almacenamiento de glucógeno tipo II también es una enfermedad metabólica. Muchos médicos no están familiarizados con los síntomas de la enfermedad de Pompe. Además, no existe una queja uniforme para la enfermedad de Pompe de aparición tardía. La enfermedad es mucho más fácil de diagnosticar en los niños.
La mayoría de los pacientes adultos ven a un médico con una variedad de problemas musculares. Los síntomas descritos pueden afectar las extremidades, pero también los músculos respiratorios o el corazón. También pueden verse afectados órganos como el hígado. El declive avanza rápidamente. Como resultado, con el tiempo, las visitas al médico aumentan sin que se realice el diagnóstico correcto. Una odisea a través de las prácticas no es infrecuente para la enfermedad de Pompe.
Terapia y tratamiento
La enfermedad de Pompe aún no se ha curado. No se dispone de un tratamiento causal de los síntomas. Por tanto, los pacientes son tratados principalmente de forma sintomática y de apoyo. Se recomiendan formas paliativas de terapia. Esta terapia incluye recomendaciones dietéticas y ejercicios de respiración, así como fisioterapia para fortalecer los músculos. En el transcurso de esto, la ventilación y la nutrición artificial se vuelven necesarias.
La decisión oportuna de estas medidas es absolutamente necesaria para prolongar la vida. En términos de nutrición, una dieta rica en proteínas en combinación con entrenamiento de resistencia ha demostrado su eficacia. Desde 2006, ha sido posible suministrar artificialmente la proteína recombinante, que consiste en células CHO y se conoce como alglucosidasa alfa o miozima. El medicamento se administra por vía intravenosa cada 14 días. Se observaron grandes éxitos en bebés después de la administración temprana del fármaco, lo que podría asegurar la supervivencia.
Hay experiencias contradictorias con niños mayores y no hay evidencia convincente de la efectividad de la forma adulta. El coste de la medicación puede ser de hasta 50.000 euros al año para un bebé y hasta 500.000 euros al año para un adulto. Es necesario un suministro de por vida. La respuesta de los músculos esqueléticos a la terapia es variable. Pero la debilidad del músculo cardíaco mejora. Debido a la barrera hematoencefálica, el fármaco no tiene ningún efecto sobre los procesos de enfermedad en el cerebro.
Los enfoques terapéuticos, como la terapia génica, se encuentran en etapas experimentales con animales. La transferencia de genes ya ha tenido éxito en ratones. El tratamiento con chaperonas farmacológicas puede aumentar la actividad residual de la maltasa ácida, pero hasta ahora no se puede utilizar de manera práctica. Se recomienda a las familias afectadas una atención psicoterapéutica de apoyo para hacer frente a la situación.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el dolorOutlook y pronóstico
La enfermedad de Pompe es una enfermedad hereditaria incurable que generalmente resulta en una esperanza de vida más corta. El pronóstico exacto depende de la forma específica de la enfermedad y de la edad de la persona afectada cuando estalla la enfermedad.
El pronóstico es peor para la forma temprana de la enfermedad de Pompe que, si no se trata, generalmente conduce a la muerte en dos años. Los afectados en su mayoría mueren de neumonía o insuficiencia cardíaca. El tratamiento de la enfermedad con terapia de reemplazo enzimático mejora significativamente el pronóstico. La esperanza de vida de los afectados aumenta significativamente hasta superar los diez años. Dado que esta forma de terapia es nueva, todavía no hay pronósticos a largo plazo.
La forma juvenil de la enfermedad de Pompe, si no se trata, generalmente conduce a la muerte antes de llegar a la edad adulta. La forma adulta de la enfermedad de Pompe tiene el pronóstico más favorable. En cualquier caso, la esperanza de vida aumenta significativamente con el tratamiento. Sin embargo, los afectados suelen desarrollar algunas limitaciones como dificultades cognitivas y pérdida de audición o incluso sordera. Actualmente se están desarrollando y probando nuevas opciones de tratamiento, como las terapias génicas para la enfermedad de Pompe, lo que debería conducir a un pronóstico mucho más favorable.
prevención
Hasta ahora, la enfermedad de Pompe solo se puede prevenir mediante el asesoramiento genético durante la fase de planificación familiar. Para los padres de un niño afectado, el riesgo de recurrencia es del 25 por ciento. Después de un diagnóstico prenatal positivo, los futuros padres también tienen la opción de un aborto.
Cura postoperatoria
Como regla general, los afectados por la enfermedad de Pompe tienen muy pocas y muy limitadas medidas y opciones de atención de seguimiento disponibles, por lo que idealmente deberían consultar a un médico en una etapa temprana para prevenir la aparición de otras quejas y complicaciones. No se puede curar de forma independiente, por lo que se debe consultar a un médico ante los primeros signos y síntomas de la enfermedad.
El proceso de recuperación se basa principalmente en ejercicios de fisioterapia o fisioterapia. Muchos de los ejercicios se pueden repetir en su propia casa, lo que favorece significativamente el tratamiento. La ayuda y el cuidado de la propia familia también es muy importante, lo que también puede prevenir la depresión y otros trastornos psicológicos.
En el caso de que exista un deseo de tener hijos, los afectados deben aprovechar las pruebas genéticas y el asesoramiento para evitar que la enfermedad vuelva a aparecer. También son necesarios controles y exámenes médicos regulares a lo largo de la vida. La enfermedad en sí no suele reducir la esperanza de vida del paciente. Por lo general, la persona interesada no dispone de más medidas de seguimiento.
Puedes hacerlo tu mismo
Para los afectados, la enfermedad de Pompe es un diagnóstico muy estresante, especialmente si la enfermedad solo se manifiesta en la vejez. La enfermedad puede tener un curso muy individual, por lo que los pacientes quieren hacer todo lo posible por un curso leve de la enfermedad.
Lo principal es prevenir enfermedades adicionales. Por ejemplo, los pacientes deben asegurarse de que están bien abastecidos de oxígeno para que no se caigan por mareos o sufran un accidente. Además, un suministro insuficiente de oxígeno dañaría el corazón y posiblemente también otros órganos. Las enfermedades infecciosas como la gripe o infecciones similares a la gripe también deben evitarse si es posible porque también afectan la respiración y debilitan gravemente el cuerpo y su sistema inmunológico. Por tanto, los pacientes con enfermedad de Pompe harían bien en prestar especial atención a su sistema inmunológico. Debe asegurarse de llevar una dieta fresca y rica en proteínas como parte de las recomendaciones dietéticas de su especialista.
El ejercicio regular también es importante para mantener la fuerza de los músculos, especialmente en las piernas. Sobre todo, el entrenamiento de resistencia ha demostrado su eficacia en el tratamiento de la enfermedad de Pompe. Los fisioterapeutas tratantes brindan la asistencia necesaria. El apoyo psicológico es útil para muchos pacientes y sus familiares. También puede obtener asistencia e información de Pompe Deutschland e.V. (www.morbus-pompe.de).
.jpg)