los Síndrome de Hippel-Lindau es una enfermedad tumoral benigna hereditaria principalmente de la retina y el cerebelo. Se basa en una malformación de los vasos sanguíneos. También pueden verse afectados otros órganos.
¿Qué es el síndrome de Hippel-Lindau?

© deagreez - stock.adobe.com
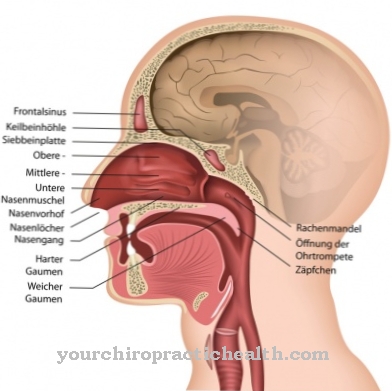
los Síndrome de Hippel-Lindau Es un cambio tisular abultado benigno muy raro, principalmente en el área de la retina y el cerebelo. Los denominados angiomas (esponjas de sangre) se presentan como tumores. Es por eso que la enfermedad a menudo se llama angiomatosis retinocerebelosa designado. El tronco encefálico y la médula espinal también suelen verse afectados.
Los tumores se desarrollan a partir de preformas del tejido conectivo y consisten en vasos sanguíneos. En su mayoría son benignos, pero también pueden degenerar maliciosamente. A veces, los tumores se encuentran en el páncreas, la glándula suprarrenal, el epidídimo o los riñones. Los tumores de riñón en particular pueden convertirse en cáncer con más frecuencia. La enfermedad recibió su nombre del oftalmólogo alemán Eugen von Hippel y del patólogo sueco Arvid Lindau. En 1904 von Hippel descubrió angiomas en la retina del ojo.
22 años después, en 1926, Arvid Lindau describió angiomas en la médula espinal. Además del nombre síndrome de Hippel-Lindau, la enfermedad también se encuentra bajo Síndrome de Von-Hippel-Lindau-Czermak, Enfermedad de Hippel-Lindau, angiomatosis retinocerebelosa o Angiomatosis retiniana conocido. Es un síndrome neurocutáneo, que se caracteriza por malformaciones vasculares en varios órganos. Los síndromes neurocutáneos son enfermedades que se manifiestan en la piel y el sistema nervioso central.
causas
El síndrome de Hippel-Lindau es genético. La enfermedad se hereda como un rasgo autosómico dominante. Sin embargo, la gravedad del síndrome también depende de muchos otros factores. Aunque el defecto genético se transmite a la siguiente generación, la gravedad de la enfermedad varía dentro de la familia. Se produce una mutación espontánea en el 50 por ciento. Entonces, la mitad de los enfermos de la familia no tienen problemas hereditarios.
Sin embargo, esto significa que varias mutaciones en el gen HL en el cromosoma 3 pueden ser responsables del desarrollo de la enfermedad. Este gen tiene una gran influencia en el desarrollo de los vasos sanguíneos y el ciclo celular. No es solo una mutación en el gen HL lo que conduce a la desregulación de los vasos sanguíneos. Ahora se ha establecido que cuando estalla la enfermedad, hay una serie de mutaciones que se distribuyen por todo el gen.
Síntomas, dolencias y signos
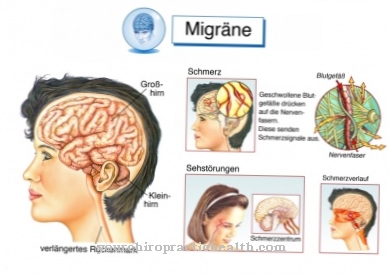
El síndrome de Hippel-Lindau generalmente se manifiesta en agotamiento, hipertensión arterial y dolores de cabeza. Además, existen síntomas neurológicos como trastornos del equilibrio, trastornos de la coordinación del movimiento o signos de presión intracraneal. Las alteraciones visuales (alteraciones visuales) suelen ser los síntomas iniciales. Sin embargo, los síntomas dependen en gran medida del tamaño y la ubicación de los tumores.
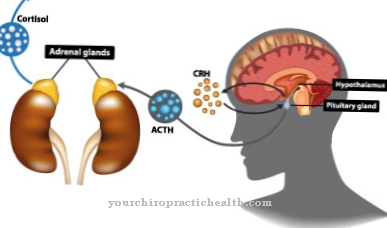
Los angiomas generalmente se encuentran en la retina, en partes del sistema nervioso central, en la médula espinal o en el tronco encefálico. El cerebro rara vez se ve afectado. Los exámenes a menudo también revelan malformaciones en otros órganos. Los quistes se encuentran especialmente en el páncreas, el hígado o los riñones. Las malformaciones vasculares arteriovenosas benignas también están presentes en el hígado. Cuando las glándulas suprarrenales tienen un angioma, se desarrolla un feocromocitoma.
Hay formas de síndrome de Hippel-Lindau con y sin feocromocitoma. El feocromocitoma es un tumor benigno de la glándula suprarrenal que produce una mayor cantidad de hormonas adrenalina y noradrenalina. Aumentan la frecuencia cardíaca y la presión arterial. La presión arterial aumenta a intervalos y reacciona con especial fuerza en situaciones de estrés.
Diagnóstico y curso de la enfermedad
La detección de múltiples hemangiomas en la retina de los ojos es una clara evidencia del síndrome de Hippel-Lindau. Un historial familiar proporciona información sobre cualquier acumulación dentro de la familia o parientes. Los procedimientos de imágenes pueden detectar posibles tumores en los riñones, las glándulas suprarrenales, el páncreas o el hígado.
Complicaciones
Varios órganos pueden verse afectados por el síndrome de Hippel-Lindau. En la mayoría de los casos, sin embargo, la persona en cuestión sufre un sentimiento general de enfermedad. Esto conduce a un fuerte dolor de cabeza y la persona afectada parece agotada. Además, se produce hipertensión arterial, que en el peor de los casos puede provocar un infarto. El paciente también se queja de problemas de visión y movilidad restringida.
La coordinación del paciente también puede verse alterada por el síndrome de Hippel-Lindau. En muchos casos, el síndrome de Hippel-Lindau también tiene un efecto negativo en el comportamiento del paciente, por lo que no es raro que se vuelva experto. La frecuencia cardíaca aumenta incluso en situaciones sencillas y fáciles, por lo que las situaciones estresantes pueden provocar sudoración o ataques de pánico para la persona en cuestión. Sin tratamiento para el síndrome de Hippel-Lindau, la esperanza de vida suele reducirse.
El síndrome no se puede tratar en todos los casos. Esto es especialmente cierto si el síndrome es genético. Sin embargo, el tratamiento sintomático puede limitar los síntomas y posiblemente extirpar el tumor. El curso exacto de la enfermedad depende de la gravedad del tumor. Por tanto, la esperanza de vida del paciente también puede ser limitada.
¿Cuándo deberías ir al médico?
En caso de una sensación general de enfermedad, malestar o fatiga, se debe realizar una estrecha autoobservación. Si la fatiga persiste a pesar de una noche de sueño reparador, es señal de un problema de salud. Si los síntomas persisten durante varias semanas, existe un motivo para un chequeo. Si los síntomas se propagan o se intensifican, se debe consultar a un médico. Consulte a un médico en caso de aumento de la presión arterial, alteración de la coordinación o secuencias de movimientos motores.
Si la presión arterial aumenta inusualmente, especialmente en situaciones estresantes de la vida, es necesaria una visita al médico. Si hay una sensación de presión dentro de la cabeza, dolores de cabeza, alteraciones visuales o una limitación del sistema auditivo, es motivo de preocupación. En casos severos, hay una pérdida auditiva completa. Esto debe examinarse y tratarse lo antes posible. Un médico debe aclarar los problemas en el área de la espalda, una disminución en el rendimiento físico y una experiencia de dolor difuso.
Si los síntomas aparecen sin motivo, la persona en cuestión necesita un examen médico. En caso de trastornos funcionales del área gastrointestinal, experiencia de calor o fluctuaciones en el estado de ánimo, se recomienda una consulta con un médico. El malestar en la región renal o las anomalías al orinar se consideran advertencias del cuerpo que deben seguirse.
Doctores y terapeutas en su área
Tratamiento y Terapia
Dado que el síndrome de Hippel-Lindau es una enfermedad genética, no es posible un tratamiento causal. Sin embargo, los angiomas existentes se pueden extirpar mediante una variedad de procedimientos. Éstos incluyen la coagulación láser, la crioterapia, la braquiterapia, la termoterapia transpupilar, la terapia fotodinámica, la radioterapia, la terapia de protones o el tratamiento con medicamentos. En la terapia con láser, los angiomas más pequeños se desnaturalizan por el sobrecalentamiento local. El tejido enfermo muere y se cura en este punto.
La crioterapia usa temperaturas tan bajas como menos 80 grados para congelar los angiomas periféricos en la retina. En la braquiterapia, la radiación radiactiva se usa para destruir los angiomas. La termoterapia transpupilar se puede utilizar para retinoblastomas, melanomas coroideos o hemangiomas coroideos. Funciona sobre la base de calentar el tumor con radiación infrarroja. En el caso de los angiomas, la presentación del éxito del tratamiento es contradictoria. Algunos estudios han informado de éxito en el tratamiento del angioma. En otros estudios, el tratamiento fue ineficaz.
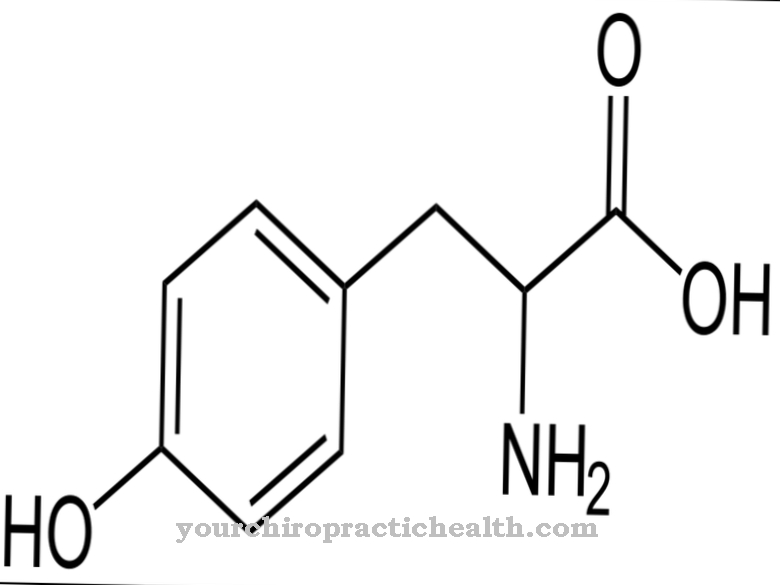
En la terapia fotodinámica, la luz se usa en combinación con una sustancia activa a la luz. La verteporfina se utiliza como sustancia activa a la luz en los estudios actuales. Se nota una mejora en la vista. Sin embargo, puede ocurrir edema macular. La radioterapia no muestra resultados significativos. Se puede mejorar la vista, pero no todos los tumores se encogen de manera uniforme. Los mejores resultados se obtienen con angiomas pequeños. La terapia de protones funciona con una precisión muy alta. Se utiliza cuando los angiomas están cerca de tejido sensible.
Outlook y pronóstico
El pronóstico del síndrome de Hippel-Lindau depende en gran medida del tipo y ubicación de los distintos tumores. Se determinó que la esperanza de vida media era de 50 años. Sin embargo, la esperanza de vida y la calidad de vida pueden aumentar significativamente mediante la detección y el tratamiento tempranos de los tumores. Aunque los tumores inicialmente simplemente deforman el tejido conectivo de los vasos sanguíneos y son benignos, algunos de ellos pueden convertirse en tumores malignos. Los carcinomas de riñón, que son la principal causa de la alta mortalidad, se desarrollan con especial frecuencia.
También se presentan carcinomas de páncreas (cáncer de páncreas) y carcinomas de otros órganos. El cáncer de páncreas es uno de los tumores particularmente agresivos que pueden conducir rápidamente a la muerte. Otra causa común de muerte es el hemangioblastoma en el cerebro, que puede provocar una hemorragia cerebral. Los síntomas generales como presión arterial alta, dolor de cabeza y fatiga, así como los síntomas neurológicos, también dependen de los tumores individuales.
Algunos pacientes no presentan síntomas si aún no han tenido hemangioblastomas en el sistema nervioso central. Los hemangioblastomas en la retina pueden provocar problemas en la retina a medida que avanza la enfermedad e incluso pueden provocar ceguera total. Además, los tumores que causan una pérdida auditiva completa son posibles en alrededor del 10 por ciento de los pacientes. El curso de la enfermedad puede ser tan diferente que no es posible un pronóstico exacto para el individuo afectado.
prevención
No existe prevención del síndrome de Hippel-Lindau porque es una enfermedad genética. Si existe una acumulación familiar de la enfermedad, se debe realizar un asesoramiento genético si el niño desea tener hijos.
Cura postoperatoria
Después de ser diagnosticado con el síndrome de Hippel-Lindau, la vida cambia para los afectados. A partir de ahora tienes que vigilar tu cuerpo con atención y consultar a un médico para aclarar cada nuevo bulto. Cuanto antes se tratan las masas, mayor es la probabilidad de un tratamiento exitoso y menos complicaciones.
Los pacientes deben someterse a chequeos periódicos durante toda su vida. Dado que pueden aparecer nuevas demandas espaciales en todo el cuerpo, estas deben ser realizadas por médicos de diferentes especialidades. En los exámenes clínicos generales anuales, se discuten todas las masas palpables, se mide la presión arterial y se discute el tratamiento adicional. Los exámenes oftalmológicos anuales permiten la detección temprana de hemangioblastomas retinianos.
La recolección de orina también se controla anualmente, que se puede usar para diagnosticar feocromocitomas. Los exámenes de resonancia magnética de la cabeza y la médula espinal se llevan a cabo cada tres años para obtener imágenes y tratar los hemangioblastomas espinales. Una resonancia magnética de abdomen también sirve para descartar feocromocitomas, carcinomas de células renales y tumores pancreáticos.
Los médicos tratantes también pueden utilizar otros métodos de examen como la tomografía por emisión de positrones o la tomografía de misión de fotón único o la gammagrafía para complementar el diagnóstico del tipo y la diseminación de los tumores. En casos individuales, puede ser necesario un examen con catéter de los vasos sanguíneos para obliterar los vasos que conducen al tumor y facilitar la terapia posterior.
Puedes hacerlo tu mismo
Con el síndrome de Hippel-Lindau, no hay posibilidades especiales de autoayuda disponibles para los afectados. Desafortunadamente, el síndrome no se puede prevenir ni tratar de manera causal, por lo que solo se administra tratamiento sintomático. Sin embargo, incluso después de un tratamiento exitoso, el paciente a menudo depende de exámenes regulares para diagnosticar y tratar otros tumores en una etapa temprana.
Los tumores generalmente se extirpan mediante cirugía. El tipo de intervención depende en gran medida de la ubicación y la gravedad del tumor. Sin embargo, como regla general, el síndrome de Hippel-Lindau no reduce la esperanza de vida de la persona afectada, ya que los tumores pueden extirparse y son benignos. A menudo, los trastornos psicológicos o la depresión se pueden evitar hablando con otras personas afectadas o con amigos cercanos y familiares. Especialmente con los niños, siempre debe tener lugar una conversación informativa para informarles sobre las posibles consecuencias de la enfermedad.
En la mayoría de los casos, el tratamiento del síndrome de Hippel-Lindau también puede mejorar la vista de la persona afectada. Sin embargo, estos todavía dependen de ayudas visuales para hacer frente a la vida cotidiana. Debido a la presión arterial alta, se deben evitar situaciones estresantes y deportes o actividades extenuantes. Esto protege la circulación y el corazón del paciente.
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)





.jpg)
.jpg)



.jpg)