Bajo la Síndrome de TAR, Inglés Síndrome de radio ausente de trombocitopenia, la medicina comprende un síndrome de malformación, cuyos principales síntomas incluyen la falla de los radios y la trombocitopenia. Se cree que la causa del síndrome es una mutación genética hereditaria. El tratamiento en los primeros años de vida consiste principalmente en transfusión de plaquetas.
¿Qué es el síndrome de TAR?
© reing - stock.adobe.com
los Síndrome de TAR es un complejo de múltiples malformaciones que se manifiesta en los recién nacidos. Los principales síntomas del síndrome de malformación hereditaria son una falla bilateral del radio y la falta de plaquetas en sangre. Debido a los síntomas, el síndrome a veces se fecha Síndrome de aplasia-trombocitopenia radial el discurso. El síndrome de TAR se ha descrito en poco más de 100 casos. Se desconoce la prevalencia exacta, pero el complejo de síntomas se considera relativamente raro.
El síndrome se describió por primera vez en 1929. Se considera que los estadounidenses H. M. Greenwald y J. Sherman son los primeros en describirlo. Según la documentación anterior, las mujeres tienen una probabilidad ligeramente mayor de verse afectadas por malformaciones que los hombres. Debido a su rareza, el síndrome no se ha investigado completamente. La investigación de la causa ha arrojado resultados parciales, pero hasta ahora no ha podido proporcionar una explicación suficiente para todo el complejo.
causas
En 2007 se identificó una posible causa del síndrome de TAR que corresponde a una mutación genética. Los síntomas parciales del complejo son causados por una microdeleción en el cromosoma 1 en el locus del gen q21.1. En este contexto, estamos hablando del síndrome de deleción 1q21.1. El cromosoma 1 se ha asociado hasta ahora con numerosas enfermedades hereditarias.
Las mutaciones en la ubicación de este gen pueden desencadenar el síndrome de Usher, la enfermedad de Gaucher o la enfermedad de Alzheimer, por ejemplo. El cromosoma 1 está presente como un par de cromosomas en todas las células del cuerpo y corresponde al cromosoma humano más grande. La mutación asociada con el síndrome de TAR parece estar necesariamente presente en todos los pacientes. Sin embargo, la mutación no explica adecuadamente los síntomas individuales del síndrome.
El síndrome de TAR se considera una enfermedad hereditaria. Se observó acumulación familiar en los casos documentados hasta el momento. La herencia parece ser un modo de herencia autosómico recesivo y una variabilidad relativamente grande en la manifestación.
Síntomas, dolencias y signos
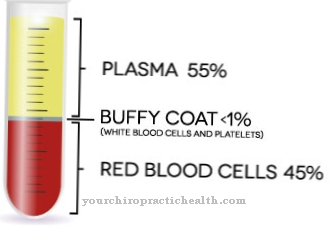
Todos los pacientes con síndrome de TAR tienen trombocitopenia. La falta de plaquetas conduce a una mayor tendencia a sangrar. Disminuyen particularmente en los primeros dos años de vida. En los primeros meses puede producirse una hemorragia intracraneal, que puede favorecer el retraso motor o mental. Bilateralmente, todos los pacientes con el síndrome también carecen de radios.
El pulgar de la persona afectada está ahí, pero solo funciona de manera anormal. A menudo hay una desviación radial de la mano, que se manifiesta como una deformidad de la mano zambo. El cúbito de todos los pacientes con TAR se acorta y se dobla parcialmente. Aproximadamente un tercio de los pacientes carece de húmero, que también suele estar acortado y tiene un efecto displásico. Las articulaciones del codo, hombro y mano están restringidas en su movilidad.
En algunos casos también hay cambios en la sangre. En dos tercios de los casos, hay un gran aumento de leucocitos. A menudo también existe una alergia o intolerancia a la leche de vaca que promueve la diarrea o agrava la trombocitopenia. En aproximadamente la mitad de todos los pacientes, los síntomas están asociados con displasia de las extremidades inferiores.
En particular, la displasia de cadera, coxa valga, subluxación de la articulación de la rodilla o displasia rotuliana con dislocación son síntomas comunes. La rodilla puede estar rígida. Las posiciones de los pies y los dedos de los pies suelen ser anormales. Muchos de los afectados también sufren de baja estatura o un defecto cardíaco en el sentido de una tetralogía de Fallot o un defecto del tabique auricular. A menudo, el ojo tiene ptosis o glaucoma.
Diagnóstico y curso de la enfermedad
En los primeros meses de vida, el médico observará una tendencia al sangrado y trombocitopenia en los pacientes con TAR, que debe diferenciar de la anemia de Fanconi en el diagnóstico diferencial. En las imágenes de rayos X, el síndrome de TAR es particularmente evidente en la falta de alineación de los radios en ambos lados y las desalineaciones resultantes.
En términos de diagnóstico diferencial, también se deben considerar el síndrome de Holt-Oram y el síndrome de Roberts. Tan pronto como han pasado los dos primeros años de vida, el pronóstico para los pacientes con síndrome de TAR es bastante favorable. En casos individuales, el pronóstico se basa en síntomas acompañantes, como el defecto cardíaco.
Complicaciones
Se producen varias malformaciones en el síndrome de TAR. En primer lugar, las malformaciones conducen a una tendencia significativamente mayor a sangrar. Los afectados sufren hemorragias graves, incluso con heridas muy leves y leves, que no se pueden detener fácilmente. El sangrado ocurre a menudo en las encías o en la nariz y tiene un efecto muy negativo en la calidad de vida de la persona afectada.
Además, el retraso mental puede ocurrir debido al síndrome TAR. En sus vidas, los pacientes a menudo dependen de la ayuda de otras personas y no pueden hacer muchas cosas cotidianas por sí mismos. La movilidad de los hombros y las manos también está significativamente restringida por el síndrome, ya que falta el húmero. Además, puede provocar un defecto cardíaco o molestias en los ojos.
El síndrome generalmente se asocia con una disminución de la esperanza de vida. Los padres o familiares también suelen sufrir trastornos psicológicos o depresión. Por lo general, no hay complicaciones con el tratamiento sintomático del síndrome de TAR. Desafortunadamente, no todas las quejas pueden restringirse por completo.
¿Cuándo deberías ir al médico?
En la mayoría de los casos, con el síndrome de TAR, la persona afectada necesitará evaluación y tratamiento médicos. No puede haber una curación independiente, por lo que la persona afectada por esta enfermedad siempre depende de un diagnóstico médico. Cuanto antes se reconozca el síndrome, mejor será el curso posterior de la enfermedad. Dado que es una enfermedad hereditaria, no puede tener lugar una curación completa. Si la persona afectada por el síndrome desea tener hijos, también se puede utilizar la asesoría genética.
En el caso del síndrome de TAR, se debe consultar a un médico si la persona en cuestión padece un retraso mental severo. Como regla general, los pacientes dependen de la ayuda de otras personas en sus vidas. La movilidad de la persona afectada también puede verse restringida por el síndrome TAR, por lo que es necesaria la visita al médico. No es raro que los órganos internos se vean afectados por varios defectos. El diagnóstico del síndrome de TAR puede realizarlo un médico de cabecera o un pediatra. Para un tratamiento adicional, es necesaria una visita a un especialista.
Terapia y tratamiento
El síndrome de TAR no puede tratarse de forma causal ni específica. Hasta ahora, solo se encuentran disponibles tratamientos sintomáticos. No se puede corregir las malformaciones en los primeros años de vida debido a la tendencia al sangrado. En etapas posteriores de la vida, las intervenciones quirúrgicas reconstructivas pueden corregir los radios faltantes y múltiples malposiciones. La prevención de todo sangrado o hemorragia es fundamental en los primeros años de vida.
El objetivo de la terapia inicial es sobre todo reducir las consecuencias importantes de la enfermedad. Las trombocitopenias graves en los primeros años de vida requieren transfusiones de plaquetas. En sí mismo no existe ningún trastorno del desarrollo motor. Las limitaciones neurológicas también son raras. El desarrollo mental pasa desapercibido. Por tanto, todos los retrasos son, en el mejor de los casos, consecuencia de hemorragias intracraneales, que deben prevenirse mediante transfusiones.
Cuando la trombocitopenia ha remitido, se llevan a cabo las medidas de tratamiento quirúrgico plástico. Estas medidas van acompañadas de [[[Pasos del tratamiento fisioterapéutico de fisioterapia]], que tienen por objeto garantizar un perfecto desarrollo motor. En la edad adulta, los pacientes a menudo ya no dependen de ninguna medida de tratamiento y llevan una vida en gran parte normal con una calidad de vida sin restricciones.
Puedes encontrar tu medicación aquí
➔ Medicamentos para el tratamiento de heridas y lesionesprevención
Las causas definitivas del síndrome de TAR aún no se han aclarado. Por esta razón, el síndrome aún no se puede prevenir. Sin embargo, todo indica que los factores genéticos juegan un papel en el síndrome. Por lo tanto, el asesoramiento genético para los afectados puede describirse en gran medida como una medida preventiva.
Cura postoperatoria
La atención de seguimiento para el síndrome de TAR depende del tipo y la gravedad de las malformaciones. Después de un procedimiento quirúrgico, que es una opción para malformaciones menores, el paciente requiere un cuidado de seguimiento extenso. Es necesaria atención inmediata en la clínica en caso de hemorragia aguda. Luego, el paciente necesita unos días de descanso.
Un examen de seguimiento final tiene como objetivo determinar las medidas de tratamiento adicionales. Las personas que padecen síndrome TAR deben consultar a su médico periódicamente para aclarar su estado de salud actual. Las complicaciones médicas no siempre son evidentes para el paciente, particularmente con hemorragia interna. En el caso de trombopenia severa, puede ser necesaria una transfusión de sangre, que se realiza en la clínica y generalmente se asocia con un debriefing.
El paciente, a su vez, necesita descanso y protección. Por lo general, está indicada la hospitalización. Los pacientes que padecen el síndrome TAR necesitan un médico especialista. El médico responsable suele ser el internista o el médico de cabecera que ya participa en el tratamiento. Se recomienda la hospitalización a largo plazo para las molestias crónicas. El paciente también debe ponerse en contacto con un fisioterapeuta y otros especialistas. El paciente también puede necesitar apoyo psicológico.
Puedes hacerlo tu mismo
El síndrome de TAR solo puede tratarse sintomáticamente. El paciente debe prestar atención a las señales de advertencia e informar al médico para que se pueda realizar una transfusión de sangre de manera temprana. Después de una transfusión de este tipo, el cuerpo se debilita y es importante garantizar una dieta equilibrada que ayude al cuerpo a producir sangre.
Después del tratamiento quirúrgico de las malformaciones, se aplica reposo y reposo en cama. El paciente debe cuidar las heridas según las indicaciones del médico para evitar inflamación y otras complicaciones. En el caso de malformaciones en las extremidades, también puede ser necesario un tratamiento fisioterapéutico. Las personas afectadas pueden hacer fisioterapia en casa y mejorar la coordinación de las extremidades afectadas con ejercicio regular.
Si estas medidas no logran el resultado deseado, se debe consultar al médico. Dado que el síndrome TAR es una afección extremadamente rara, un médico especialista debe hacerse cargo de la terapia. Es aconsejable buscar en foros de Internet para otros pacientes, ya que solo hay unos pocos grupos de autoayuda para la afección. Finalmente, es importante realizar las visitas necesarias al médico para evitar complicaciones graves. El síndrome de TAR debe ser monitoreado de cerca debido a las transfusiones de sangre.
.jpg)













.jpg)

.jpg)
.jpg)